
Xeljanz
Ultimo aggiornamento: 12/02/2024
Cos'è Xeljanz?
Xeljanz è un farmaco a base del principio attivo
Tofacitinib, appartenente alla categoria degli
Farmaci per il trattamento dell'artrite reumatoide e nello specifico
. E' commercializzato in Italia dall'azienda
Pfizer S.r.l..
Xeljanz può essere prescritto con Ricetta RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti.
Xeljanz può essere prescritto con Ricetta RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti.
Confezioni
Xeljanz 10 mg 112 compresse rivestite con film
Xeljanz 5 mg 112 compresse a rilascio prolungato
Xeljanz 5 mg 182 compresse rivestite con film
Xeljanz 5 mg 56 compresse rivestite con film
Xeljanz 5 mg 112 compresse a rilascio prolungato
Xeljanz 5 mg 182 compresse rivestite con film
Xeljanz 5 mg 56 compresse rivestite con film
Informazioni commerciali sulla prescrizione
Titolare: Pfizer Europe MA EEIG
Concessionario: Pfizer S.r.l.
Ricetta: RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti
Classe: H
Principio attivo: Tofacitinib
Gruppo terapeutico: Farmaci per il trattamento dell'artrite reumatoide
ATC: L04AF01 - Tofacitinib
Forma farmaceutica: compresse rivestite
Concessionario: Pfizer S.r.l.
Ricetta: RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti
Classe: H
Principio attivo: Tofacitinib
Gruppo terapeutico: Farmaci per il trattamento dell'artrite reumatoide
ATC: L04AF01 - Tofacitinib
Forma farmaceutica: compresse rivestite
Se sei un professionista, potrai trovare le schede tecniche complete e molto altro nell'area riservata di Codifa.it
Indicazioni
Perché si usa Xeljanz? A cosa serve?
Artrite reumatoide
Tofacitinib in associazione con metotrexato (MTX) è indicato per il trattamento dell'artrite reumatoide (AR) in fase attiva da moderata a severa in pazienti adulti che hanno risposto in modo inadeguato o sono intolleranti ad uno o più farmaci antireumatici modificanti la malattia (DMARD) (vedere paragrafo 5.1).
Tofacitinib può essere somministrato in monoterapia in caso di intolleranza a MTX o quando il trattamento con MTX non è appropriato (vedere paragrafi 4.4 e 4.5).
Artrite psoriasica
Tofacitinib in associazione a MTX è indicato per il trattamento dell'artrite psoriasica attiva (PsA) in pazienti adulti che hanno risposto in modo inadeguato o sono intolleranti ad una precedente terapia con un farmaco antireumatico modificante la malattia (DMARD) (vedere paragrafo 5.1).
Tofacitinib è indicato per il trattamento di pazienti adulti affetti da spondilite anchilosante (SA) attiva che hanno risposto in modo inadeguato alla terapia convenzionale.
Colite ulcerosa
Tofacitinib è indicato per il trattamento di pazienti adulti affetti da colite ulcerosa (CU) attiva da moderata a severa che hanno manifestato una risposta inadeguata, hanno perso la risposta o sono intolleranti alla terapia convenzionale o a un agente biologico (vedere paragrafo 5.1).
Artrite idiopatica giovanile (JIA)
Tofacitinib è indicato per il trattamento dell'artrite idiopatica giovanile poliarticolare in fase attiva (poliartrite con fattore reumatoide positivo [RF+] o negativo [RF-] e oligoartrite estesa) e dell'artrite psoriasica (PsA) giovanile in pazienti di età pari o superiore a 2 anni, che hanno risposto in modo inadeguato alla precedente terapia con DMARD.
Tofacitinib può essere somministrato in associazione con metotrexato (MTX) o in monoterapia in caso di intolleranza a MTX o quando sia inappropriato continuare un trattamento con MTX.
Posologia
Come usare Xeljanz: Posologia
Il trattamento deve essere iniziato e seguito da medici specialisti con esperienza nella diagnosi e nel trattamento delle condizioni per le quali è indicato Tofacitinib.
Posologia
La dose raccomandata è compresse rivestite con film da 5 mg somministrate due volte al giorno. Tale dose non deve essere superata.
Non è richiesta alcuna modifica di dose quando usato in associazione a MTX
Tabella 1: Passaggio da tofacitinib compresse rivestite con film a tofacitinib compresse a rilascio prolungato, e viceversa
|
Passaggio da tofacitinib 5 mg compresse rivestite con film a tofacitinib 11 mg compresse a rilascio prolungato e viceversaa
|
Il trattamento con tofacitinib 5 mg compresse rivestite con film due volte al giorno e il trattamento con tofacitinib 11 mg compresse a rilascio prolungato una volta al giorno possono essere scambiati tra loro il giorno successivo all'ultima dose di uno dei trattamenti.
|
a Vedere paragrafo 5.2 per il confronto della farmacocinetica delle formulazioni a rilascio prolungato e rivestite con film.
Spondilite anchilosanteLa dose raccomandata di tofacitinib è di 5 mg somministrati due volte al giorno.
Colite ulcerosa
Trattamento di induzione
La dose raccomandata è di 10 mg somministrati per via orale due volte al giorno per l'induzione per 8 settimane.
Per i pazienti che non raggiungono un adeguato beneficio terapeutico entro la settimana 8, la dose di induzione di 10 mg due volte al giorno può essere prorogata di altre 8 settimane (16 settimane totali), proseguendo con 5 mg due volte al giorno per il mantenimento. La terapia di induzione con tofacitinib deve essere interrotta in tutti i pazienti che non manifestano beneficio terapeutico entro la settimana 16.
Trattamento di mantenimento
La dose raccomandata per il trattamento di mantenimento è tofacitinib 5 mg somministrato per via orale due volte al giorno.
Tofacitinib 10 mg due volte al giorno per il trattamento di mantenimento non è raccomandato nei pazienti affetti da CU che presentano fattori di rischio noti per tromboembolismo venoso (TEV), eventi avversi cardiovascolari maggiori (MACE) e tumori maligni, a meno che non esista alcun trattamento alternativo adeguato (vedere paragrafi 4.4 e 4.8).
Nei pazienti affetti da CU che non presentano un rischio aumentato di TEV, MACE e tumori maligni (vedere paragrafo 4.4), può essere preso in considerazione tofacitinib 10 mg per via orale due volte al giorno se il paziente manifesta una riduzione della risposta a tofacitinib 5 mg due volte al giorno e non ha risposto a opzioni di trattamento alternative per la colite ulcerosa, come il trattamento con un inibitore del fattore di necrosi tumorale (inibitore del TNF). Tofacitinib 10 mg due volte al giorno per il trattamento di mantenimento deve essere usato per la durata più breve possibile. Deve essere somministrata la dose più bassa possibile che sia efficace per il mantenimento della risposta.
Nei pazienti che hanno risposto al trattamento con tofacitinib, è possibile ridurre e/o sospendere l'assunzione di corticosteroidi secondo lo standard di cura.
Se la terapia viene interrotta, è possibile prendere in considerazione la ripresa del trattamento con tofacitinib. Se si è verificata una perdita di risposta, è possibile prendere in considerazione la reinduzione con tofacitinib 10 mg due volte al giorno. Il periodo di interruzione del trattamento negli studi clinici è stato prorogato sino a 1 anno. L'efficacia può essere recuperata con 8 settimane di terapia con 10 mg due volte al giorno (vedere paragrafo 5.1).
JIA poliarticolare e PsA giovanile (bambini di età compresa tra 2 e 18 anni)
Tofacitinib può essere somministrato in monoterapia o in associazione a MTX.
La dose raccomandata nei pazienti di età pari o superiore a 2 anni si basa sulle seguenti categorie di peso:
Tabella 2: Dose di tofacitinib per pazienti con artrite idiopatica giovanile poliarticolare e PsA giovanile di età pari o superiore a due anni
|
Peso corporeo (kg)
|
Posologia
|
|
10 - < 20
|
3,2 mg (3,2 mL di soluzione orale) due volte al giorno
|
|
20 - < 40
|
4 mg (4 mL di soluzione orale) due volte al giorno
|
|
≥ 40
|
5 mg (5 mL di soluzione orale o 5 mg compressa rivestita con film) due volte al giorno
|
I pazienti che pesano ≥ 40 kg trattati con tofacitinib 5 mL soluzione orale due volte al giorno possono passare a tofacitinib 5 mg compresse rivestite con film due volte al giorno. I pazienti che pesano < 40 kg non possono abbandonare la soluzione orale di tofacitinib per passare alle compresse rivestite con film.
Sospensione e interruzione del trattamento negli adulti e nei pazienti pediatrici
Il trattamento con tofacitinib deve essere sospeso se un paziente sviluppa un'infezione grave fino a quando l'infezione non si sia risolta.
Può essere necessaria l'interruzione del trattamento per la gestione delle alterazioni risultanti dagli esami di laboratorio relative alla dose, tra cui linfopenia, neutropenia e anemia. Come descritto nelle Tabelle 3, 4 e 5 sottostanti, le raccomandazioni per una interruzione temporanea o permanente del trattamento sono fatte in relazione alla severità delle alterazioni risultanti dagli esami di laboratorio (vedere paragrafo 4.4).
Si raccomanda di non iniziare il trattamento nei pazienti con una conta linfocitaria assoluta (ALC) inferiore a 750 cellule/mm3.
Tabella 3: Bassa conta linfocitaria assoluta
|
Bassa conta linfocitaria assoluta (ALC) (vedere paragrafo 4.4)
|
|
|
Valore di laboratorio
(cellule/mm3)
|
Raccomandazione
|
|
ALC maggiore o uguale a 750
|
La dose deve essere mantenuta.
|
|
ALC 500-750
|
Per riduzioni persistenti in questo intervallo (2 valori consecutivi in questo intervallo agli esami di routine) la somministrazione deve essere ridotta o sospesa.
Per i pazienti che ricevono tofacitinib 10 mg due volte al giorno, il dosaggio deve essere ridotto a tofacitinib 5 mg due volte al giorno.
Per i pazienti che ricevono tofacitinib 5 mg due volte al giorno, la somministrazione deve essere interrotta.
Quando l'ALC è maggiore di 750, il trattamento deve essere ripreso in base alle indicazioni cliniche.
|
|
ALC minore di 500
|
Se il valore di laboratorio è confermato da un test ripetuto entro 7 giorni, la somministrazione deve essere interrotta.
|
Si raccomanda di non iniziare il trattamento nei pazienti adulti con una conta assoluta dei neutrofili (ANC – Absolute Neutrophil Count) inferiore a 1.000 cellule/mm3. Si raccomanda di non iniziare il trattamento nei pazienti pediatrici con una conta assoluta dei neutrofili (ANC – Absolute Neutrophil Count) inferiore a 1.200 cellule/mm3.
Tabella 4: Bassa conta assoluta dei neutrofili
|
Bassa conta assoluta dei neutrofili (ANC) (vedere paragrafo 4.4)
|
|
|
Valore di laboratorio
(cellule/mm3)
|
Raccomandazione
|
|
ANC maggiore di 1.000
|
La dose deve essere mantenuta.
|
|
ANC 500-1.000
|
Per riduzioni persistenti in questo intervallo (2 valori consecutivi in questo intervallo agli esami di routine), la somministrazione deve essere ridotta o sospesa.
Per i pazienti che ricevono tofacitinib 10 mg due volte al giorno, il dosaggio deve essere ridotto a tofacitinib 5 mg due volte al giorno.
Per i pazienti che ricevono tofacitinib 5 mg due volte al giorno, la somministrazione deve essere interrotta.
Quando l'ANC è maggiore di 1.000, il trattamento deve essere ripreso in base alle indicazioni cliniche.
|
|
ANC inferiore a 500
|
Se il valore di laboratorio è confermato da un test ripetuto entro 7 giorni, la somministrazione deve essere interrotta.
|
Si raccomanda di non iniziare il trattamento nei pazienti adulti con emoglobina inferiore a 9 g/dL. Si raccomanda di non iniziare il trattamento nei pazienti pediatrici con emoglobina inferiore a 10 g/dL.
Tabella 5: Basso valore dell'emoglobina
|
Basso valore dell'emoglobina (vedere paragrafo 4.4)
|
|
|
Valore di laboratorio (g/dL)
|
Raccomandazione
|
|
Riduzione inferiore o uguale a 2 g/dL e valore superiore o uguale a 9,0 g/dL
|
La dose deve essere mantenuta.
|
|
Riduzione superiore a 2 g/dL o valore inferiore a 8,0 g/dL (confermata da test ripetuti)
|
La somministrazione deve essere sospesa fino a quando i valori di emoglobina non si siano normalizzati.
|
Interazioni
La dose totale giornaliera di tofacitinib deve essere dimezzata nei pazienti che ricevono i potenti inibitori del citocromo P450 (CYP) 3A4 (ad es. ketoconazolo) e nei pazienti che ricevono 1 o più medicinali concomitanti che provocano sia una moderata inibizione di CYP3A4 sia una potente inibizione di CYP2C19 (ad es. fluconazolo) (vedere paragrafo 4.5) come segue:
- La dose di tofacitinib deve essere ridotta a 5 mg una volta al giorno in pazienti che assumono 5 mg due volte al giorno (pazienti adulti e pediatrici).
- La dose di tofacitinib deve essere ridotta a 5 mg due volte al giorno in pazienti che assumono 10 mg due volte al giorno (pazienti adulti).
Solo nei pazienti pediatrici:
i dati disponibili suggeriscono che un miglioramento clinico si osserva in 18 settimane dall'inizio del trattamento con tofacitinib.. La prosecuzione della terapia deve essere attentamente riconsiderata in un paziente che non presenta miglioramenti clinici entro tale periodo.
I dati disponibili suggeriscono che un miglioramento clinico si osserva nella SA entro 16 settimane dall'inizio del trattamento con tofacitinib. La prosecuzione della terapia deve essere attentamente riconsiderata in un paziente che non presenta miglioramenti clinici entro tale periodo.
Popolazioni speciali
Anziani
Non è necessaria alcuna modifica di dose in pazienti di età pari o superiore ai 65 anni. Sono disponibili dati limitati in pazienti di età pari o superiore ai 75 anni. Vedere paragrafo 4.4 per l'Uso in pazienti di età pari o superiore a 65 anni.
Compromissione epatica
Tabella 6: Modifica di dose per compromissione epatica
|
Categoria di compromissione epatica
|
Classificazione
|
Modifica di dose nella compromissione epatica per compresse a dosaggio diverso
|
|
Lieve
|
Child Pugh A
|
Non è necessaria alcuna modifica di dose.
|
|
Moderata
|
Child Pugh B
|
La dose deve essere ridotta a 5 mg una volta al giorno quando la dose indicata in presenza di una normale funzionalità epatica è di 5 mg due volte al giorno.
La dose deve essere ridotta a 5 mg due volte al giorno quando la dose indicata in presenza di una normale funzionalità epatica è di 10 mg due volte al giorno (vedere paragrafo 5.2).
|
|
Severa
|
Child Pugh C
|
Tofacitinib non deve essere usato in pazienti affetti da severa compromissione epatica (vedere paragrafo 4.3).
|
Compromissione renale
Tabella 7: Modifica di dose per compromissione renale
|
Categoria di compromissione renale
|
Clearance della creatinina
|
Modifica di dose nella compromissione renale per compresse a dosaggio diverso
|
|
Lieve
|
50-80 mL/min
|
Non è necessaria alcuna modifica di dose.
|
|
Moderata
|
30-49 mL/min
|
Non è necessaria alcuna modifica di dose.
|
|
Severa (compresi i pazienti sottoposti a emodialisi)
|
<30 mL/min
|
La dose deve essere ridotta a 5 mg una volta al giorno quando la dose indicata in presenza di una normale funzionalità renale è di 5 mg due volte al giorno.
La dose deve essere ridotta a 5 mg due volte al giorno quando la dose indicata in presenza di una normale funzionalità renale è di 10 mg due volte al giorno.
I pazienti affetti da compromissione renale severa devono mantenere una dose ridotta anche dopo l'emodialisi (vedere paragrafo 5.2).
|
Popolazione pediatrica
La sicurezza e l'efficacia di tofacitinib nei bambini di età inferiore a 2 anni con JIA poliarticolare e PsA giovanile non sono state stabilite. Non ci sono dati disponibili.
La sicurezza e l'efficacia di tofacitinib nei bambini di età inferiore a 18 anni con altre indicazioni (ad es. colite ulcerosa) non sono state stabilite. Non ci sono dati disponibili.
Modo di somministrazione
Uso orale.
Tofacitinib viene somministrato per via orale con o senza cibo.
Per i pazienti che hanno difficoltà nella deglutizione, le compresse di tofacitinib possono essere frantumate e assunte con acqua.
Controindicazioni
Quando non dev'essere usato Xeljanz
- Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
- Tubercolosi (TB) attiva, infezioni gravi come sepsi o infezioni opportunistiche (vedere paragrafo 4.4).
- Compromissione epatica severa (vedere paragrafo 4.2).
- Gravidanza e allattamento (vedere paragrafo 4.6).
Avvertenze speciali e precauzioni di impiego
Cosa serve sapere prima di prendere Xeljanz
Tofacitinib deve essere utilizzato solo se non sono disponibili alternative terapeutiche adeguate nei pazienti:
- di età pari o superiore a 65 anni;
- con anamnesi di malattia cardiovascolare aterosclerotica o altri fattori di rischio cardiovascolare (come i pazienti fumatori da lungo tempo o coloro che per lungo tempo hanno fumato in passato);
- con fattori di rischio per tumori maligni (ad es. tumore maligno in corso o anamnesi di tumore maligno).
Uso in pazienti di età pari o superiore a 65 anni
In considerazione dell'aumento del rischio di infezioni gravi, infarto miocardico, tumori maligni e mortalità per tutte le cause con tofacitinib in pazienti di età pari o superiore a 65 anni, tofacitinib deve essere usato in questi pazienti solo nel caso in cui non siano disponibili alternative terapeutiche adeguate (vedere ulteriori dettagli di seguito nei paragrafi 4.4 e 5.1).
Associazione con altre terapie
Tofacitinib non è stato studiato e deve essere evitato in associazione a farmaci biologici, come antagonisti del TNF, antagonisti dell'interleuchina (IL)-1R, antagonisti dell'IL-6R, anticorpi monoclonali anti-CD20, antagonisti dell'IL-17, antagonisti dell'IL-12/IL-23, anti-integrine, modulatori selettivi della co-stimolazione e immunosoppressori potenti, come azatioprina, 6-mercaptopurina, ciclosporina e tacrolimus, a causa del possibile aumento dell'immunosoppressione e dell'aumentato rischio di infezione.
Negli studi clinici sull'AR, vi è stata un'incidenza maggiore di eventi avversi per l'associazione di tofacitinib a MTX, rispetto a tofacitinib in monoterapia.
L'uso di tofacitinib in associazione a inibitori della fosfodiesterasi 4 non è stato esaminato negli studi clinici su tofacitinib.
Tromboembolismo venoso (TEV)
Eventi gravi di TEV, tra cui embolia polmonare (EP), alcuni dei quali fatali, e trombosi venosa profonda (TVP), sono stati osservati in pazienti che assumevano tofacitinib. In uno studio randomizzato di sicurezza successivo all'autorizzazione condotto in pazienti con artrite reumatoide di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, è stato osservato un aumento del rischio di TEV dose-dipendente con tofacitinib rispetto agli inibitori del TNF (vedere paragrafi 4.8 e 5.1).
In un'analisi esplorativa a posteriori (post hoc) condotta nell'ambito di questo studio, in pazienti con fattori di rischio di TEV noti, insorgenze di TEV successive sono state osservate più frequentemente nei pazienti trattati con tofacitinib che, a 12 mesi di trattamento, presentavano un livello di D-dimero ≥ 2 volte l'ULN, rispetto a quelli con livello di D-dimero < 2 volte l'ULN; ciò non è stato rilevato nei pazienti trattati con inibitori del TNF. L'interpretazione è limitata dal basso numero di eventi di TEV e dalla disponibilità limitata del test del D-dimero (valutato solo al basale, al mese 12 e alla fine dello studio). Nei pazienti che non hanno manifestato un TEV durante lo studio, i livelli medi di D-dimero erano significativamente ridotti al mese 12 rispetto al basale in tutti i bracci di trattamento. Tuttavia, sono stati osservati livelli di D-dimero ≥ 2 volte l'ULN al mese 12 in circa il 30% dei pazienti senza successivi eventi di TEV, a indicare una specificità limitata del test del D-dimero in questo studio.
Tofacitinib 10 mg due volte al giorno per il trattamento di mantenimento non è raccomandato nei pazienti affetti da CU che presentano fattori di rischio noti di TEV, MACE e tumori maligni, a meno che non esista alcun trattamento alternativo adeguato (vedere paragrafo 4.2).
Nei pazienti con fattori di rischio cardiovascolare o per tumori maligni (vedere anche paragrafo 4.4 “Eventi avversi cardiovascolari maggiori (MACE)” e “Tumori maligni”), tofacitinib deve essere utilizzato solo se non sono disponibili alternative terapeutiche adeguate.
Nei pazienti con fattori di rischio di TEV diversi dai fattori di rischio di MACE o tumori maligni, tofacitinib deve essere usato con cautela. I fattori di rischio di TEV diversi dai fattori di rischio di MACE o tumori maligni comprendono TEV precedente, pazienti sottoposti a un intervento chirurgico maggiore, immobilizzazione, uso di contraccettivi ormonali combinati o terapia ormonale sostitutiva, disturbo ereditario della coagulazione. Durante il trattamento con tofacitinib, i pazienti devono essere sottoposti periodicamente a nuova valutazione per determinare eventuali variazioni del rischio di TEV.
Per i pazienti affetti da artrite reumatoide con fattori di rischio di TEV noti, prendere in considerazione di analizzare i livelli di D-dimero dopo circa 12 mesi di trattamento. Qualora il risultato del test del D-dimero sia ≥ 2 volte l'ULN, accertarsi che i benefici clinici siano superiori ai rischi prima di prendere una decisione in merito alla prosecuzione del trattamento con tofacitinib.
Valutare tempestivamente i pazienti con segni e sintomi di TEV e interrompere la somministrazione di tofacitinib nei pazienti con sospetto TEV, indipendentemente dalla dose o dall'indicazione.
Trombosi venosa retinica
Nei pazienti in trattamento con tofacitinib è stata riportata trombosi venosa retinica (TVR) (vedere paragrafo 4.8). I pazienti devono essere avvisati di rivolgersi immediatamente a un medico nel caso in cui manifestino sintomi indicativi di TVR.
Infezioni gravi
Sono state riportate infezioni gravi e talvolta fatali, causate da batteri, micobatteri, funghi invasivi, virus o altri patogeni opportunisti, in pazienti in trattamento con tofacitinib (vedere paragrafo 4.8). Il rischio di infezioni opportunistiche è più alto nelle regioni geografiche dell'Asia (vedere paragrafo 4.8). I pazienti affetti da artrite reumatoide che assumono corticosteroidi possono essere predisposti a infezioni.
Tofacitinib non deve essere iniziato in pazienti con infezioni attive, incluse le infezioni localizzate.
Devono essere considerati i rischi e i benefici del trattamento prima di iniziare tofacitinib in pazienti:
- con infezioni ricorrenti,
- con una anamnesi di infezione grave o opportunistica,
- che hanno vissuto o viaggiato in aree di micosi endemica,
- che hanno condizioni di base che possano predisporre all'infezione,
I pazienti devono essere attentamente monitorati per lo sviluppo di segni e sintomi di infezione durante e dopo il trattamento con tofacitinib. Il trattamento deve essere interrotto se un paziente sviluppa un'infezione grave, un'infezione opportunistica o sepsi. Un paziente che sviluppa una nuova infezione durante il trattamento con tofacitinib deve essere sottoposto ad esami diagnostici tempestivi e completi, adeguati al paziente immunocompromesso, deve essere avviata un'appropriata terapia antibiotica e il paziente deve essere attentamente monitorato.
Poiché vi è una maggiore incidenza di infezioni negli anziani e nella popolazione diabetica in generale, si raccomanda cautela nel trattamento degli anziani e dei pazienti diabetici (vedere paragrafo 4.8). Nei pazienti di età pari o superiore ai 65 anni, tofacitinib deve essere utilizzato solo se non sono disponibili alternative terapeutiche adeguate (vedere paragrafo 5.1).
Il rischio di infezione può essere più alto con gradi maggiori di linfopenia e deve essere considerata la conta linfocitaria nel valutare il rischio di infezione dei singoli pazienti. I criteri di interruzione e monitoraggio per la linfopenia sono analizzati nel paragrafo 4.2.
Tubercolosi
Devono essere considerati i rischi e i benefici del trattamento prima di iniziare tofacitinib in pazienti:
- che sono stati esposti a TB,
- che hanno vissuto o viaggiato in aree di TB endemica.
I pazienti devono essere valutati ed esaminati per infezione latente o attiva prima e, secondo le linee guida applicabili, durante la somministrazione di tofacitinib.
I pazienti con TB latente, risultati positivi al test, devono essere trattati con terapia antimicobatterica standard prima della somministrazione di tofacitinib.
La terapia antitubercolare deve essere anche considerata prima della somministrazione di tofacitinib in pazienti che risultino negativi al test per TB, ma che abbiano una pregressa anamnesi di TB latente o attiva, e ove non possa essere confermato un adeguato percorso terapeutico; oppure per i pazienti risultati negativi al test, ma che presentano fattori di rischio per l'infezione tubercolare. Si raccomanda il consulto di un medico con esperienza nel trattamento della TB per decidere se sia opportuno iniziare la terapia antitubercolare per ogni singolo paziente. I pazienti devono essere attentamente monitorati per lo sviluppo di segni e sintomi di TB, compresi i pazienti che sono risultati negativi al test per l'infezione tubercolare latente prima di iniziare la terapia.
Riattivazione virale
Nei pazienti trattati con tofacitinib, l'incidenza di herpes zoster sembra essere aumentata in:
- Pazienti giapponesi o coreani;
- Pazienti con un'ALC inferiore a 1.000 cellule/mm3 (vedere paragrafo 4.2);
- Pazienti con AR di lunga durata, che hanno assunto precedentemente due o più farmaci antireumatici modificanti la malattia (DMARD) biologici.
- Pazienti trattati con 10 mg due volte al giorno.
Non è noto l'effetto di tofacitinib sulla riattivazione dell'epatite virale cronica. I pazienti con screening positivo per l'epatite B o C sono stati esclusi dagli studi clinici. Lo screening per l'epatite virale deve essere eseguito in conformità con le linee guida cliniche prima di iniziare la terapia con tofacitinib.
Eventi avversi cardiovascolari maggiori (tra cui infarto del miocardio)
Sono stati osservati eventi avversi cardiovascolari maggiori (MACE) in pazienti che assumevano tofacitinib.
In uno studio randomizzato di sicurezza successivo all'autorizzazione condotto in pazienti con artrite reumatoide di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, è stata osservata una maggiore incidenza di infarti del miocardio con tofacitinib rispetto agli inibitori del TNF (vedere paragrafi 4.8 e 5.1). Nei pazienti di età pari o superiore a 65 anni, nei pazienti fumatori da lungo tempo o coloro che per lungo tempo hanno fumato in passato e nei pazienti con anamnesi di malattia cardiovascolare aterosclerotica o altri fattori di rischio cardiovascolare, tofacitinib deve essere usato solo nel caso in cui non siano disponibili alternative terapeutiche adeguate (vedere paragrafo 5.1).
Tumori maligni e disordini linfoproliferativi
Sono stati osservati NMSC, cancro del polmone e linfoma in pazienti trattati con tofacitinib anche in altri studi clinici e nella fase successiva all'immissione in commercio.
Sono stati osservati altri tumori maligni in pazienti trattati con tofacitinib in studi clinici e nella fase post-commercializzazione, tra cui, ma non solo, cancro al seno, melanoma, cancro della prostata e cancro del pancreas.
Nei pazienti di età pari o superiore a 65 anni, nei pazienti fumatori da lungo tempo o coloro che per lungo tempo hanno fumato in passato e nei pazienti con altri fattori di rischio di tumore maligno (ad esempio, tumore maligno in corso o anamnesi di tumore maligno escluso il cancro della cute non melanoma trattato con successo), tofacitinib deve essere usato solo nel caso in cui non siano disponibili alternative terapeutiche adeguate (vedere paragrafo 5.1). Esami cutanei periodici sono raccomandati per tutti i pazienti, in particolare per coloro che presentano un rischio maggiore di tumore cutaneo (vedere Tabella 8 nel paragrafo 4.8).
Malattia polmonare interstiziale
Si raccomanda cautela anche nei pazienti con una anamnesi di malattia polmonare cronica, in quanto tali pazienti possono essere più soggetti alle infezioni. Casi di malattia polmonare interstiziale (alcuni dei quali hanno avuto un esito fatale) sono stati riportati in pazienti affetti da AR trattati con tofacitinib negli studi clinici e nella fase successiva alla commercializzazione, sebbene il ruolo dell'inibizione della Janus chinasi (JAK) in questi eventi non sia noto. È noto che i pazienti asiatici affetti da artrite reumatoide sono a più alto rischio di malattia polmonare interstiziale, quindi si deve usare cautela nel trattamento di questi pazienti.
Perforazioni gastrointestinali
Negli studi clinici sono stati riportati eventi di perforazione gastrointestinale, sebbene il ruolo dell'inibizione di JAK in questi eventi non sia noto.
Tofacitinib deve essere usato con cautela nei pazienti che possono presentare un aumentato rischio di perforazione gastrointestinale (ad esempio, pazienti con una anamnesi di diverticolite, pazienti in trattamento con corticosteroidi e/o farmaci antinfiammatori non steroidei). I pazienti che presentano segni e sintomi addominali di nuova insorgenza devono essere valutati tempestivamente per l'individuazione precoce di perforazione gastrointestinale.
Fratture
In pazienti trattati con tofacitinib sono stati osservati casi di fratture.
Tofacitinib deve essere usato con cautela in pazienti con noti fattori di rischio di fratture quali pazienti anziani, pazienti di sesso femminile e pazienti in trattamento con corticosteroidi, indipendentemente dall'indicazione e dal dosaggio.
Enzimi epatici
Il trattamento con tofacitinib è stato associato ad un aumento dell'incidenza di innalzamento degli enzimi epatici in alcuni pazienti (vedere test degli enzimi epatici, paragrafo 4.8). Si deve usare cautela nel considerare l'inizio del trattamento con tofacitinib in pazienti con elevati livelli di alanina aminotransferasi (ALT) o aspartato aminotransferasi (AST), in particolare quando si inizia il trattamento in associazione a medicinali potenzialmente epatotossici, come il MTX. Dopo l'inizio del trattamento, si consiglia il monitoraggio di routine dei test epatici e una tempestiva indagine sulle cause di eventuali innalzamenti degli enzimi epatici osservati, al fine di identificare potenziali casi di traumatismo epatico indotto dal farmaco. Se si sospetta un traumatismo epatico farmaco-indotto, la somministrazione di tofacitinib deve essere interrotta fino a quando non sia stata esclusa questa diagnosi.
Ipersensibilità
Nell'esperienza post marketing sono stati segnalati casi di ipersensibilità associati alla somministrazione di tofacitinib. Le reazioni allergiche includevano angioedema e orticaria; si sono verificate reazioni gravi. Se si verifica una reazione allergica o anafilattica grave, il tofacitinib deve essere sospeso immediatamente.
Parametri di laboratorio
Linfociti
Il trattamento con tofacitinib è stato associato ad un aumento dell'incidenza di linfopenia rispetto al placebo. Una conta linfocitaria inferiore a 750 cellule/mm3 è stata associata ad un aumento dell'incidenza di infezioni gravi. Si raccomanda di non iniziare o continuare il trattamento con tofacitinib nei pazienti con una conta linfocitaria confermata inferiore a 750 cellule/mm3. I linfociti devono essere monitorati al basale e ogni 3 mesi successivi. Per le modifiche raccomandate sulla base della conta linfocitaria, vedere paragrafo 4.2.
Neutrofili
Il trattamento con tofacitinib è stato associato ad un aumento dell'incidenza di neutropenia (meno di 2.000 cellule/mm3) rispetto al placebo. Si raccomanda di non iniziare il trattamento con tofacitinib nei pazienti adulti con un ANC inferiore a 1.000 cellule/mm3 e nei pazienti pediatrici con un ANC inferiore a 1.200 cellule/mm3. L'ANC deve essere monitorata al basale, dopo 4-8 settimane di trattamento e, successivamente, ogni 3 mesi. Per le modifiche raccomandate in base all'ANC, vedere paragrafo 4.2.
Emoglobina
Il trattamento con tofacitinib è stato associato a riduzione dei livelli di emoglobina. È raccomandato di non iniziare il trattamento con tofacitinib nei pazienti adulti con un valore di emoglobina inferiore a 9 g/dL e nei pazienti pediatrici con un valore di emoglobina inferiore a 10 g/dL. L'emoglobina deve essere monitorata al basale, dopo 4-8 settimane di trattamento e, successivamente, ogni 3 mesi. Per le modifiche raccomandate in base al livello di emoglobina, vedere paragrafo 4.2.
Monitoraggio dei lipidi
Il trattamento con tofacitinib è stato associato ad aumenti dei parametri lipidici, quali colesterolo totale, colesterolo LDL (lipoproteine a bassa densità) e colesterolo HDL (lipoproteine ad alta densità). Effetti massimi sono stati osservati generalmente entro 6 settimane. La valutazione dei parametri lipidici deve essere eseguita dopo 8 settimane dall'inizio della terapia con tofacitinib. I pazienti devono essere trattati secondo le linee guida cliniche per la gestione dell'iperlipidemia. Gli aumenti del colesterolo totale e LDL associati a tofacitinib possono essere ridotti ai livelli pre-trattamento con statine.
Ipoglicemia nei pazienti in trattamento per il diabete
Sono stati segnalati casi di ipoglicemia dopo l'inizio del trattamento con tofacitinib in pazienti che assumevano farmaci per il diabete. In caso di ipoglicemia può essere necessaria una modifica della dose dei farmaci antidiabetici.
Vaccinazioni
Prima di iniziare il trattamento con tofacitinib, si raccomanda che tutti i pazienti, in particolare i pazienti affetti da JIA e jPsA, abbiano completato tutte le vaccinazioni, in accordo con le linee guida vigenti sull'immunizzazione. Si raccomanda di non somministrare vaccini vivi in concomitanza con tofacitinib. La decisione di utilizzare vaccini vivi prima del trattamento con tofacitinib deve tener conto dell'immunosoppressione preesistente di un dato paziente.
In base alle linee guida sulla vaccinazione, deve essere presa in considerazione la vaccinazione profilattica per zoster. Va prestata particolare attenzione ai pazienti con AR di lunga durata che hanno assunto precedentemente due o più DMARD biologici. Se viene somministrato vaccino vivo per lo zoster, deve essere somministrato solo a pazienti con una storia nota di varicella o a pazienti sieropositivi per il virus della varicella zoster (VZV). Se l'anamnesi di varicella è incerta o inattendibile si raccomanda di testare gli anticorpi contro VZV.
La vaccinazione con vaccini vivi deve essere eseguita almeno 2 settimane prima, ma preferibilmente 4 settimane prima di iniziare tofacitinib o in conformità con le linee guida attuali sulla vaccinazione in merito ai medicinali immunomodulanti. Non sono disponibili dati sulla trasmissione secondaria di infezione da vaccini vivi a pazienti che ricevono tofacitinib.
Eccipienti contenuti:
Questo medicinale contiene lattosio. I pazienti affetti da rari problemi ereditari di intolleranza al galattosio, da deficit totale di lattasi o da malassorbimento di glucosio-galattosio non devono assumere questo medicinale.
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per compressa. Cioè si può definire essenzialmente “privo di sodio”.
Interazioni con altri medicinali e altre forme di interazione
Quali farmaci o alimenti possono modificare l'effetto di Xeljanz
Possibilità da parte di altri medicinali di influenzare la farmacocinetica (PK) di tofacitinib
Poiché tofacitinib è metabolizzato da CYP3A4, è probabile che si verifichi l'interazione con medicinali che inibiscono o inducono CYP3A4. L'esposizione a tofacitinib è aumentata quando co-somministrato con potenti inibitori di CYP3A4 (ad esempio, ketoconazolo)o quando la co-somministrazione di uno o più medicinali determina sia l'inibizione moderata di CYP3A4, sia l'inibizione potente di CYP2C19 (ad esempio, fluconazolo)(vedere paragrafo 4.2).
L'esposizione a tofacitinib è ridotta quando co-somministrato con potenti induttori del CYP (ad esempio, rifampicina). È improbabile che gli inibitori del solo CYP2C19 o della glicoproteina-P alterino significativamente la PK di tofacitinib.
La co-somministrazione con ketoconazolo (forte inibitore di CYP3A4), fluconazolo (inibitore moderato di CYP3A4 e potente di CYP2C19), tacrolimus (inibitore lieve di CYP3A4) e ciclosporina (inibitore moderato di CYP3A4) ha aumentato l'AUC di tofacitinib, mentre la rifampicina (potente induttore di CYP) ha diminuito l'AUC di tofacitinib. La co-somministrazione di tofacitinib con potenti induttori di CYP (ad esempio, rifampicina) può comportare una riduzione o la perdita della risposta clinica (vedere Figura 1). Si raccomanda di non co-somministrare tofacitinib con potenti induttori di CYP3A4. La co-somministrazione con ketoconazolo e fluconazolo ha aumentato la Cmax di tofacitinib, mentre tacrolimus, ciclosporina e rifampicina hanno ridotto la Cmax di tofacitinib. La co-somministrazione di MTX 15-25 mg una volta alla settimana non ha avuto alcun effetto sulla PK di tofacitinib in pazienti affetti da AR (vedere Figura 1).
Figura 1. Impatto di altri medicinali sulla PK di tofacitinib
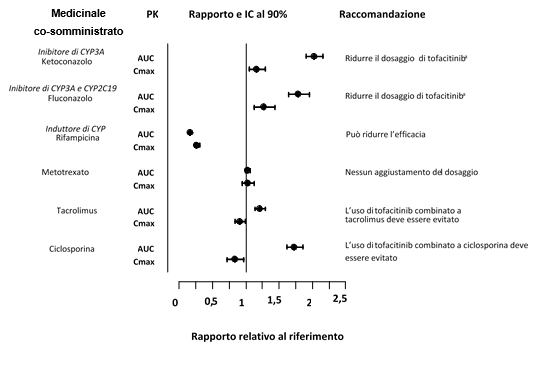
Nota: il gruppo di riferimento è la somministrazione di tofacitinib in monoterapia.
a La dose di tofacitinib deve essere ridotta a 5 mg due volte al giorno in pazienti che assumono 10 mg due volte al giorno. La dose di di tofacitinib deve essere ridotta a 5 mg una volta al giorno in pazienti che assumono 5 mg due volte al giorno (vedere paragrafo 4.2).
Possibilità di tofacitinib di influenzare la PK di altri medicinali
La co-somministrazione di tofacitinib in volontarie sane non ha avuto effetti sulla PK di contraccettivi orali, levonorgestrel ed etinilestradiolo.
In pazienti con AR, la co-somministrazione di tofacitinib con MTX 15-25 mg una volta alla settimana ha diminuito l'AUC e la Cmax di MTX del 10% e 13%, rispettivamente. L'entità della riduzione dell'esposizione a MTX non giustifica modifiche al dosaggio personalizzato di MTX.
Popolazione pediatrica
Sono stati effettuati studi d'interazione solo negli adulti.
Interazioni riportate su letteratura scientifica internazionale
Prima di prendere "Xeljanz" insieme ad altri farmaci come “Adoport”, “Advagraf”, “Aurantin - Soluzione (uso Interno)”, “BCG-Medac”, “Carbamazepina EG”, “Carbamazepina Zentiva - Compressa A Rilascio Modificato”, “Carelimus”, “Ciqorin”, “Conferoport”, “Dintoina”, “Dintoinale”, “Envarsus”, “Erleada”, “Fenitoina Hikma”, “Fenobarbitale Sodico Salf”, “Firacrono”, “Fluenz Tetra”, “Gamibetal Complex”, “Gardenale”, “Ikervis”, “Luminale - Compressa”, “Luminale - Soluzione (uso Interno)”, “Lysodren”, “M-M-Rvaxpro”, “Mysoline”, “Nervaxon”, “Oncotice”, “Priorix Tetra”, “Priorix”, “Prograf - Capsula”, “Prograf - Soluzione (uso Interno)”, “Proquad”, “Protopic - Unguento”, “Qdenga”, “Rifadin”, “Rifater”, “Rifinah”, “Rifocin”, “Rotarix”, “Rotateq”, “Sandimmun - Capsula Molle”, “Sandimmun Neoral - Capsula Molle”, “Sandimmun Neoral - Soluzione”, “Sandimmun - Soluzione”, “Sandimmun - Soluzione (uso Interno)”, “Stadmycin”, “Stamaril”, “Tacforius”, “Tacni”, “Tacrolimus EG”, “Tegretol”, “Varilrix”, “Varivax”, “Vaxchora”, “Verkazia”, “Xtandi”, “Zostavax”, etc.., chiedi al tuo al tuo medico o farmacista di fiducia di verificare che sia sicuro e non dannoso per la tua salute ...
Fertilità, gravidanza e allattamento
Gravidanza
Non sono disponibili studi adeguati e ben controllati sull'uso di Tofacitinib in donne in gravidanza. È stato dimostrato che tofacitinib è teratogeno in ratti e conigli e incide sul parto e sullo sviluppo peri/postnatale (vedere paragrafo 5.3).
Come misura precauzionale, l'uso di tofacitinib in gravidanza è controindicato (vedere paragrafo 4.3).
Donne in età fertile/contraccezione nelle donne
Le donne in età fertile devono essere informate della necessità di utilizzare un metodo contraccettivo efficace durante il trattamento con tofacitinib e per almeno 4 settimane dopo l'ultima dose.
Allattamento
Non è noto se tofacitinib sia secreto nel latte materno. Non si può escludere un rischio per il lattante. Tofacitinib è stato secreto nel latte di ratti in allattamento (vedere paragrafo 5.3). Come misura precauzionale, l'uso di tofacitinib durante l'allattamento al seno è controindicato (vedere paragrafo 4.3).
Fertilità
Non sono stati condotti studi specifici sul potenziale effetto sulla fertilità umana.
Nei ratti tofacitinib ha ridotto la fertilità femminile, ma non la fertilità maschile (vedere paragrafo 5.3).
Effetti sulla capacità di guidare veicoli e sull'uso di macchinari
Tofacitinib non altera o altera in modo trascurabile la capacità di guidare veicoli e di usare macchinari.
Effetti indesiderati
Quali sono gli effetti collaterali di Xeljanz
Riepilogo del profilo di sicurezza
Artrite reumatoide
Le reazioni indesiderate gravi più comuni sono state infezioni gravi (vedere paragrafo 4.4).
Nella popolazione totale esposta per la sicurezza a lungo termine, le infezioni gravi più comuni riportate con Tofacitinib sono state polmonite (1,7%), herpes zoster (0,6%), infezione delle vie urinarie (0,4%), cellulite (0,4%), diverticolite (0,3%) e appendicite (0,2%). Tra le infezioni opportunistiche, sono state segnalate con tofacitinib infezioni TB e altre infezioni da micobatteri, criptococco, istoplasmosi, candidosi esofagea, herpes zoster multidermatomerico, infezione da citomegalovirus, infezioni da virus BK e listeriosi. Alcuni pazienti hanno manifestato una malattia disseminata piuttosto che localizzata. Possono inoltre verificarsi altre infezioni gravi che non sono state riportate negli studi clinici (ad esempio, coccidioidomicosi).
Le reazioni indesiderate più comunemente riportate durante i primi 3 mesi in studi clinici in doppio cieco, controllati con placebo o MTX sono stati cefalea (3,9%), infezioni delle vie aeree superiori (3,8%), infezione virale delle vie aeree superiori (3,3%), diarrea (2,9%), nausea (2,7%) e ipertensione (2,2%).
La percentuale di pazienti che hanno interrotto il trattamento a causa di reazioni indesiderate durante i primi 3 mesi degli studi in doppio cieco, controllati con placebo o con MTX, è stata del 3,8% per i pazienti che assumevano tofacitinib. Le infezioni più comuni che hanno determinato l'interruzione della terapia nei primi 3 mesi negli studi clinici controllati sono state herpes zoster (0,19%) e polmonite (0,15%).
Nel complesso, il profilo di sicurezza osservato nei pazienti con PsA attiva trattati con tofacitinib è risultato coerente con il profilo di sicurezza osservato nei pazienti con AR trattati con tofacitinib.
Nel complesso, il profilo di sicurezza osservato nei pazienti affetti da SA attiva trattati con tofacitinib è risultato coerente con il profilo di sicurezza osservato nei pazienti affetti da AR trattati con tofacitinib.
Colite ulcerosa
Le reazioni avverse riportate più comunemente nei pazienti che hanno ricevuto tofacitinib 10 mg due volte al giorno negli studi di induzione sono state cefalea, nasofaringite, nausea e artralgia.
Negli studi di induzione e mantenimento, in tutti i gruppi di trattamento con tofacitinib e placebo, le categorie più comuni di reazioni avverse gravi sono state patologie e infezioni gastrointestinali e la reazione avversa grave più comune è stata il peggioramento della CU.
Complessivamente, il profilo di sicurezza osservato nei pazienti affetti da CU trattati con tofacitinib era coerente con il profilo di sicurezza di tofacitinib nell'indicazione dell'AR.
Tabella delle reazioni indesiderate
Le reazioni indesiderate elencate nella tabella sottostante provengono da studi clinici su pazienti affetti da AR,PsA, SA e CU e vengono presentate in base alla Classificazione per sistemi e organi (SOC - System Organ Class) e le categorie di frequenza, definita utilizzando la seguente convenzione: molto comune (≥1/10); comune (≥1/100, <1/10), non comune (≥1/1.000, <1/100), raro (≥1/10.000, <1/1.000), molto raro (<1/10.000) o non nota (non può essere definita sulla base dei dati disponibili). All'interno di ciascuna classe di frequenza, le reazioni indesiderate sono riportate in ordine decrescente di gravità.
|
Classificazione per sistemi e organi
|
Comune
≥ 1/100, < 1/10
|
Non comune
≥ 1/1.000, < 1/100
|
Raro
≥ 1/10.000, < 1/1.000
|
Molto raro
< 1/10.000
|
Non nota (non può essere definita sulla base dei dati disponibili)
|
|
Infezioni ed infestazioni
|
Polmonite
Influenza
Herpes zoster
Infezione delle vie urinarie
Sinusite
Bronchite
Nasofaringite
Faringite
|
Tubercolosi
Diverticolite
Pielonefrite
Cellulite
Herpes simplex
Gastroenterite virale
Infezione virale
|
Sepsi
Urosepsi
Tubercolosi disseminata
Batteriemia
Polmonite da Pneumocystis jirovecii
Polmonite pneumococcica
Polmonite batterica
Infezione da citomegalovirus
Artrite batterica
|
Tubercolosi del sistema nervoso centrale
Meningite criptococcica
Fascite necrotizzante
Encefalite
Batteriemia stafilococcica
Infezione da Mycobacterium avium complex Infezione da micobatteri atipici
|
|
|
Tumori benigni, maligni e non specificati (cisti e polipi compresi)
|
|
Cancro del polmone
Tumori cutanei non-melanoma
|
Linfoma
|
|
|
|
Patologie del sistema emolinfopoietico
|
Linfopenia
Anemia
|
Leucopenia
Neutropenia
|
|
|
|
|
Disturbi del sistema immunitario
|
|
|
|
|
Ipersensibilità *
Angioedema *
Orticaria *
|
|
Disturbi del metabolismo e della nutrizione
|
|
Dislipidemia
Iperlipidemia
Disidratazione
|
|
|
|
|
Disturbi psichiatrici
|
|
Insonnia
|
|
|
|
|
Patologie del sistema nervoso
|
Cefalea
|
Parestesia
|
|
|
|
|
Disturbi cardiaci
|
|
Infarto del miocardio
|
|
|
|
|
Patologie vascolari
|
Ipertensione
|
Tromboembolismo venoso**
|
|
|
|
|
Patologie respiratorie, toraciche e mediastiniche
|
Tosse
|
Dispnea
Congestione sinusale
|
|
|
|
|
Patologie gastrointestinali
|
Dolore addominale
Vomito
Diarrea
Nausea
Gastrite
Dispepsia
|
|
|
|
|
|
Patologie epatobiliari
|
|
Steatosi epatica
Aumento degli enzimi epatici
Aumento delle transaminasi
Aumento della gamma glutamil-transferasi
|
Alterazione dei test di funzionalità epatica
|
|
|
|
Patologie della cute e del tessuto sottocutaneo
|
Eruzione cutanea
Acne
|
Eritema
Prurito
|
|
|
|
|
Patologie del sistema muscoloscheletrico e del tessuto connettivo
|
Artralgia
|
Tumefazione articolare
Tendinite
|
Dolore muscoloscheletrico
|
|
|
|
Patologie sistemiche e condizioni relative alla sede di somministrazione
|
Edema periferico
|
Piressia
Stanchezza
|
|
|
|
|
Esami diagnostici
|
Aumento della creatinfosfochinasi ematica
|
Aumento della
creatinina ematica
Aumento della colesterolemia
Aumento delle lipoproteine a bassa densità
Aumento di peso
|
|
|
|
|
Traumatismo, avvelenamento e complicazioni da procedura
|
|
Distorsione di legamento
Strappo muscolare
|
|
|
|
* Segnalazioni spontanee
** Il tromboembolismo venoso comprende EP, TVP e trombosi venosa retinica
Descrizione di reazioni indesiderate specifiche
Tromboembolismo venoso
Artrite reumatoide
In un ampio studio (N=4.362) randomizzato di sicurezza successivo all'autorizzazione in pazienti con artrite reumatoide di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare (CV) aggiuntivo, è stata osservata TEV con un'incidenza aumentata e dose-dipendente nei pazienti trattati con tofacitinib rispetto a quelli trattati con inibitori del TNF (vedere paragrafo 5.1). La maggior parte di questi eventi è risultata grave e alcuni hanno causato la morte. I tassi di incidenza (IC al 95%) di EP per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF sono stati rispettivamente di 0,17 (0,08-0,33), 0,50 (0,32-0,74) e 0,06 (0,01-0,17) pazienti con eventi per 100 pazienti-anno. Rispetto agli inibitori del TNF, l'hazard ratio (HR) per l'EP è risultato rispettivamente pari a 2,93 (0,79-10,83) e 8,26 (2,49-27,43) per tofacitinib 5 mg due volte al giorno e tofacitinib 10 mg due volte al giorno (vedere paragrafo 5.1). La maggioranza (97%) dei pazienti trattati con tofacitinib in cui è stata osservata EP presentava fattori di rischio di TEV.
Negli studi clinici controllati randomizzati combinati di Fase 2 e 3, non si sono verificati eventi di TEV in 420 pazienti (233 pazienti-anno di osservazione) trattati con tofacitinib fino a 48 settimane.
Colite ulcerosa (CU)
Nello studio di estensione sulla CU in corso, sono stati osservati casi di EP e TVP in pazienti trattati con tofacitinib 10 mg due volte al giorno e con fattore(i) di rischio di TEV sottostante(i).
Infezioni generali
Artrite reumatoide
In studi clinici di fase 3 controllati, i tassi di infezioni oltre 0-3 mesi nei gruppi di tofacitinib in monoterapia 5 mg due volte al giorno (616 pazienti totali)e 10 mg due volte al giorno (642 pazienti totali)sono stati rispettivamente del 16,2% (100 pazienti) e del 17,9% (115 pazienti), paragonati al 18,9% (23 pazienti) nel gruppo placebo (122 pazienti totali). In studi clinici di fase 3 controllati con DMARD di fondo, i tassi di infezioni oltre 0-3 mesi nel gruppo tofacitinib più DMARD 5 mg due volte al giorno (973 pazienti totali)e 10 mg due volte al giorno (969 pazienti totali) sono stati rispettivamente del 21,3% (207 pazienti) e del 21,8% (211 pazienti), in confronto al 18,4% (103 pazienti) nel gruppo placebo più DMARD (559 pazienti totali).
Le infezioni più comunemente riportate sono state infezioni delle vie aeree superiori e nasofaringite (3,7% e 3,2%, rispettivamente).
Il tasso di incidenza globale delle infezioni con tofacitinib nella popolazione totale esposta per la sicurezza a lungo termine (4.867 pazienti totali) è stata di 46,1 pazienti con eventi per 100 pazienti-anno (43,8 e 47,2 pazienti con eventi per 5 mg e 10 mg due volte al giorno, rispettivamente). Per i pazienti (1.750 totali) in monoterapia, i tassi sono stati 48,9 e 41,9 pazienti con eventi per 100 pazienti-anno per 5 mg e 10 mg due volte al giorno, rispettivamente. Per i pazienti (3.117 totali) con DMARD di base, i tassi sono stati 41,0 e 50,3 pazienti con eventi per 100 pazienti-anno per 5 mg e 10 mg due volte al giorno, rispettivamente.
Negli studi clinici combinati di Fase 2 e 3, durante il periodo controllato con placebo fino a 16 settimane, la frequenza delle infezioni nel gruppo che assumeva tofacitinib 5 mg due volte al giorno (185 pazienti) è stata del 27,6% e la frequenza nel gruppo placebo (187 pazienti) è stata del 23,0%. Negli studi clinici combinati di Fase 2 e 3, tra i 316 pazienti trattati con tofacitinib 5 mg due volte al giorno fino a 48 settimane, la frequenza delle infezioni è stata del 35,1%.
Colite ulcerosa
Negli studi di induzione randomizzati di fase 2/3 a 8 settimane, le percentuali di pazienti con infezioni sono state del 21,1% (198 pazienti) nel gruppo che assumeva tofacitinib 10 mg due volte al giorno rispetto al 15,2% (43 pazienti) nel gruppo placebo. Nello studio randomizzato di mantenimento di fase 3 a 52 settimane, la percentuale di pazienti con infezioni è stata del 35,9% (71 pazienti) nel gruppo che assumeva tofacitinib 5 mg due volte al giorno e del 39,8% (78 pazienti) nel gruppo che assumeva tofacitinib 10 mg due volte al giorno, rispetto al 24,2% (48 pazienti) nel gruppo placebo.
Durante l'intera durata del trattamento con tofacitinib, l'infezione più comunemente riportata è stata la nasofaringite, che si è verificata nel 18,2% dei pazienti (211 pazienti).
Durante l'intera durata del trattamento con tofacitinib, il tasso di incidenza complessivo delle infezioni era di 60,3 eventi per 100 anni paziente (che coinvolgono il 49,4% dei pazienti, per un totale di 572 pazienti).
Infezioni gravi
Artrite reumatoide
Negli studi clinici controllati della durata di 6 mesi e 24 mesi, il tasso di infezioni gravi nel gruppo di tofacitinib in monoterapia 5 mg due volte al giorno, è stato di 1,7 pazienti con eventi per 100 pazienti-anno. Nel gruppo di tofacitinib in monoterapia 10 mg due volte al giorno, il tasso è stato di 1,6 pazienti con eventi per 100 pazienti-anno, di 0 eventi per 100 pazienti-anno per il gruppo placebo e di 1,9 pazienti con eventi per 100 pazienti-anno per il gruppo MTX.
Negli studi della durata di 6, 12 o 24 mesi, i tassi di infezioni gravi nei gruppi di tofacitinib 5 mg due volte al giorno e 10 mg due volte al giorno, associato a DMARD, sono stati rispettivamente di 3,6 e 3,4 pazienti con eventi per 100 pazienti-anno, in confronto a 1,7 pazienti con eventi per 100 pazienti-anno nel gruppo placebo più DMARD.
Nella popolazione totale esposta per la sicurezza a lungo termine, i tassi globali di infezioni gravi sono stati di 2,4 e 3,0 pazienti con eventi per 100 pazienti-anno per i gruppi di tofacitinib 5 mg e 10 mg due volte al giorno, rispettivamente. Le infezioni gravi più comuni comprendono polmonite, herpes zoster, infezione delle vie urinarie, cellulite, gastroenterite e diverticolite. Sono stati riportati casi di infezioni opportunistiche (vedere paragrafo 4.4).
I tassi di incidenza (IC al 95%) di infezioni gravi per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF sono stati rispettivamente di 2,86 (2,41; 3,37), 3,64 (3,11; 4,23) e 2,44 (2,02; 2,92) pazienti con eventi per 100 pazienti-anno. Rispetto agli inibitori del TNF, l'hazard ratio (HR) per le infezioni gravi è risultato rispettivamente pari a 1,17 (0,92; 1,50) e 1,48 (1,17; 1,87) per tofacitinib 10 mg due volte al giorno e tofacitinib 5 mg due volte al giorno.
Spondilite anchilosante
Negli studi clinici combinati di Fase 2 e 3, tra i 316 pazienti trattati con 5 mg di tofacitinib due volte al giorno fino a 48 settimane, si è verificata un'infezione grave (meningite asettica), con un tasso di incidenza di 0,43 pazienti con eventi per 100 pazienti-anno.
Colite ulcerosa
I tassi di incidenza e i tipi di infezioni gravi negli studi clinici sulla CU sono stati generalmente simili a quelli riportati negli studi clinici sull'AR in gruppi trattati in monoterapia con tofacitinib.
Infezioni gravi negli anziani
Dei 4.271 pazienti arruolati negli Studi I-VI (vedere paragrafo 5.1) sull'AR, 608 pazienti affetti da AR in totale avevano un'età pari o superiore a 65 anni, tra cui 85 pazienti con età pari o superiore a 75 anni. La frequenza di infezioni gravi tra i pazienti trattati con tofacitinib di età pari o superiore a 65 anni è stata superiore rispetto a quella tra pazienti di età inferiore a 65 anni (4,8 per 100 pazienti-anno rispetto a 2,4 per 100 pazienti-anno).
In un ampio studio (N=4.362) randomizzato post-autorizzazione sulla sicurezza condotto in pazienti con AR di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, è stato osservato un aumento delle infezioni gravi in pazienti di età pari o superiore a 65 anni trattati con tofacitinib 10 mg due volte al giorno rispetto ai pazienti trattati con inibitori del TNF e tofacitinib 5 mg due volte al giorno (vedere paragrafo 4.4). I tassi di incidenza (IC al 95%) per le infezioni gravi in pazienti ≥65 anni sono stati rispettivamente 4,03 (3,02; 5,27), 5,85 (4,64; 7,30) e 3,73 (2,81; 4,85) pazienti con eventi per 100 pazienti-anno per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF.
Rispetto agli inibitori del TNF, l'hazard ratio (HR) per le infezioni gravi in pazienti di età ≥65 anni è risultato rispettivamente pari a 1,08 (0,74; 1,58) e 1,55 (1,10; 2,19) per tofacitinib 5 mg due volte al giorno e tofacitinib 10 mg due volte al giorno.
Infezioni gravi in uno studio post-autorizzativo non interventistico sulla sicurezza
I dati ottenuti nell'ambito di uno studio post-approvazione non interventistico sulla sicurezza volto a valutare tofacitinib in pazienti con artrite reumatoide da un registro (Corrona statunitense) hanno mostrato che è stato osservato un tasso di incidenza di infezioni gravi numericamente superiore per la compressa rivestita a rilascio prolungato da 11 mg somministrata una volta al giorno rispetto alla compressa rivestita con film da 5 mg somministrata due volte al giorno. I tassi grezzi di incidenza (IC al 95%) (per esempio non aggiustati per età o sesso), dalla disponibilità di ciascuna formulazione a 12 mesi dopo l'inizio del trattamento, sono stati di 3,45 (1,93; 5,69) e 2,78 (1,74; 4,21) e, a 36 mesi, di 4,71 (3,08; 6,91) e 2,79 (2,01; 3,77) pazienti con eventi per 100 pazienti-anno per il gruppo trattato con compresse a rilascio prolungato da 11 mg una volta al giorno e il gruppo trattato con compresse rivestite con film da 5 mg due volte al giorno, rispettivamente. Il rapporto di rischio (hazard ratio) non aggiustato è stato di 1,30 (IC al 95%: 0,67; 2,50) a 12 mesi e 1,93 (IC al 95%: 1,15; 3,24) a 36 mesi per la formulazione a rilascio prolungato da 11 mg una volta al giorno rispetto alla formulazione rivestita con film da 5 mg due volte al giorno. I dati si basano su un numero ridotto di pazienti con eventi osservati con intervalli di confidenza relativamente ampi e un follow-up limitato.
Riattivazione virale
I pazienti trattati con tofacitinib giapponesi o coreani o i pazienti con AR di lunga durata, che hanno assunto precedentemente due o più DMARD biologici o i pazienti con una ALC inferiore a 1.000 cellule/mm3 o i pazienti trattati con 10 mg due volte al giorno possono presentare un rischio aumentato di herpes zoster (vedere paragrafo 4.4).
In un ampio studio (N=4.362) randomizzato di sicurezza, successivo all'autorizzazione, condotto in pazienti con AR di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, è stato osservato un aumento degli eventi di herpes zoster nei pazienti trattati con tofacitinib rispetto agli inibitori del TNF. I tassi di incidenza (IC al 95%) di herpes zoster per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF sono stati rispettivamente di 3,75 (3,22; 4,34), 3,94 (3,38; 4,57) e 1,18 (0,90; 1,52) pazienti con eventi per 100 pazienti-anno
Test di laboratorio
Linfociti
Negli studi clinici controllati sull'AR, si sono verificate diminuzioni confermate della ALC al di sotto di 500 cellule/mm3 nello 0,3% dei pazienti, e valori di ALC compresi tra 500 e 750 cellule/mm3 nell'1,9% dei pazienti, per le dosi combinate di 5 mg due volte al giorno e 10 mg due volte al giorno.
Nella popolazione con AR di sicurezza a lungo termine, si sono verificate diminuzioni confermate della ALC al di sotto di 500 cellule/mm3 nell'1,3% dei pazienti, e valori di ALC compresi tra 500 e 750 cellule/mm3 nell'8,4% dei pazienti, per le dosi combinate di 5 mg due volte al giorno e 10 mg due volte al giorno.
ALC confermate inferiori a 750 cellule/mm3 sono state associate ad un aumento dell'incidenza di infezioni gravi (vedere paragrafo 4.4).
Negli studi clinici sulla CU, le variazioni di ALC osservate con il trattamento con tofacitinib erano simili alle variazioni osservate negli studi clinici sull'AR.
Neutrofili
Negli studi clinici controllati sull'AR, si sono verificate diminuzioni confermate nell'ANC al di sotto di 1.000 cellule/mm3 nello 0,08% dei pazienti per le dosi combinate di 5 mg due volte al giorno e 10 mg due volte al giorno. Non sono state osservate diminuzioni confermate dell'ANC al di sotto di 500 cellule/mm3 in nessun gruppo di trattamento. Non è stata individuata una relazione chiara tra la neutropenia e la comparsa di infezioni gravi.
Nella popolazione con AR di sicurezza a lungo termine, l'andamento e l'incidenza delle diminuzioni confermate nella ANC sono rimasti in linea con quanto è stato osservato negli studi clinici controllati (vedere paragrafo 4.4).
Negli studi clinici sulla CU, le variazioni di ANC osservate con il trattamento con tofacitinib erano simili alle variazioni osservate negli studi clinici sull'AR.
Per essere idonei all'arruolamento i pazienti negli studi clinici controllati di Fase 3 (AR, PsA, SA, CU) dovevano presentare una conta piastrinica ≥ 100.000 cellule/mm3, pertanto non sono disponibili informazioni per i pazienti con conta piastrinica < 100.000 cellule/mm3 prima di iniziare il trattamento con tofacitinib.
Test degli enzimi epatici
Nei pazienti con AR sono stati osservati raramente aumenti confermati degli enzimi epatici maggiori di 3 volte il limite superiore del valore normale (3x ULN). In tali pazienti che hanno presentato un aumento degli enzimi epatici, una modifica del regime di trattamento, come la riduzione della dose di DMARD concomitante, l'interruzione di tofacitinib o la riduzione della dose di tofacitinib, ha portato alla diminuzione o alla normalizzazione degli enzimi epatici.
Nella parte controllata dello studio di fase 3 in monoterapia (0-3 mesi) sull'AR, (studio I, vedere paragrafo 5.1), sono stati osservati innalzamenti dell'ALT superiori a 3 volte l'ULN nell'1,65%, 0,41% e 0% dei pazienti trattati rispettivamente con placebo, tofacitinib5 mg e 10 mg due volte al giorno. In questo studio, sono stati osservati innalzamenti dell'AST superiori a 3 volte l'ULN nell'1,65%, 0,41% e 0% dei pazienti trattati rispettivamente con placebo, tofacitinib 5 mg e 10 mg due volte al giorno.
Nello studio di fase 3 in monoterapia (0-24 mesi) sull'AR, (studio VI, vedere paragrafo 5.1), sono stati osservati innalzamenti dell'ALT superiori a 3 volte l'ULN nel 7,1%, 3,0% e 3,0% dei pazienti trattati rispettivamente con MTX, tofacitinib 5 mg e 10 mg due volte al giorno. In questo studio, sono stati osservati innalzamenti dell'AST superiori a 3 volte l'ULN nel 3,3%, 1,6% e 1,5% dei pazienti trattati rispettivamente con MTX, tofacitinib 5 mg e 10 mg due volte al giorno.
Nella parte controllata degli studi di fase 3 sull'AR con DMARD di base (0-3 mesi) (Studio II-V, vedere paragrafo 5.1), sono stati osservati innalzamenti dell'ALT superiori a 3 volte l'ULN nello 0,9%, 1,24% e 1,14% dei pazienti trattati rispettivamente con placebo, tofacitinib 5 mg e 10 mg due volte al giorno. In questi studi, sono stati osservati innalzamenti dell'AST superiori a 3 volte l'ULN nello 0,72%, 0,5% e 0,31% dei pazienti trattati rispettivamente con placebo, tofacitinib 5 mg e 10 mg due volte al giorno.
Negli studi di estensione a lungo termine sull'AR, in monoterapia, sono stati osservati innalzamenti dell'ALT superiori a 3 volte l'ULN nell'1,1% e 1,4% dei pazienti trattati rispettivamente con tofacitinib 5 mg e 10 mg due volte al giorno. Innalzamenti dell'AST superiori a 3 volte l'ULN sono stati osservati in <1,0% in entrambi i gruppi tofacitinib 5 mg e 10 mg due volte al giorno.
Negli studi di estensione a lungo termine sull'AR con DMARD di fondo sono stati osservati innalzamenti dell'ALT superiori a 3 volte l'ULN nell'1,8% e 1,6% dei pazienti trattati rispettivamente con tofacitinib 5 mg e 10 mg due volte al giorno. Innalzamenti dell'AST superiori a 3 volte l'ULN sono stati osservati in < 1,0% in entrambi i gruppi tofacitinib5 mg e 10 mg due volte al giorno.
In un ampio studio (N=4.362) randomizzato di sicurezza, successivo all'autorizzazione, condotto in pazienti con AR di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, sono stati osservati innalzamenti dell'ALT superiori o uguali a 3 volte l'ULN nel 6,01%, 6,54% e 3,77% dei pazienti trattati rispettivamente con tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF. Innalzamenti dell'AST superiori o uguali a 3 volte l'ULN sono stati osservati nel 3,21%, 4,57% e 2,38% dei pazienti che ricevevano rispettivamente tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF.
Negli studi clinici sulla CU, le variazioni nei test degli enzimi epatici osservate con il trattamento con tofacitinib erano simili alle variazioni osservate negli studi clinici sull'AR.
Lipidi
Gli innalzamenti dei parametri lipidici (colesterolo totale, colesterolo LDL, colesterolo HDL, trigliceridi) sono stati valutati inizialmente 1 mese dopo l'inizio di tofacitinib negli studi clinici controllati in doppio cieco di AR. In questo punto temporale sono stati osservati aumenti, successivamente stabilizzati.
Di seguito sono riepilogate le variazioni dei parametri lipidici rispetto al basale, fino alla fine dello studio (6-24 mesi) negli studi clinici controllati in AR:
- Il colesterolo LDL medio è aumentato del 15% nel braccio di tofacitinib 5 mg due volte al giorno e del 20% nel braccio di tofacitinib10 mg due volte al giorno al mese 12, ed è aumentato del 16% nel braccio di tofacitinib5 mg due volte al giorno e del 19% nel braccio di tofacitinib 10 mg due volte al giorno al mese 24.
- Il colesterolo HDL medio è aumentato del 17% nel braccio di tofacitinib 5 mg due volte al giorno e del 18% nel braccio di tofacitinib 10 mg due volte al giorno al mese 12, ed è aumentato del 19% nel braccio di tofacitinib 5 mg due volte al giorno e del 20% nel braccio di tofacitinib10 mg due volte al giorno al mese 24.
Al momento dell'interruzione del trattamento con tofacitinib, i livelli di lipidi sono tornati ai valori basali.
I rapporti di colesterolo LDL/colesterolo HDL medi e i rapporti di apolipoproteina B (ApoB)/ApoA1 sono rimasti sostanzialmente invariati nei pazienti trattati con tofacitinib.
In uno studio clinico controllato sull'AR, l'innalzamento di colesterolo LDL e ApoB è sceso ai livelli pretrattamento in risposta alla terapia con statine.
Nelle popolazioni con AR di sicurezza a lungo termine, l'innalzamento dei parametri lipidici è rimasto in linea con ciò che è stato osservato negli studi clinici controllati.
In un ampio studio (N=4.362) randomizzato di sicurezza, successivo all'autorizzazione, condotto in pazienti con AR di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, le variazioni dei parametri lipidici dal basale a 24 mesi sono riassunte di seguito:
- Il colesterolo LDL medio è aumentato del 13,80%, 17,04% e 5,50% nei pazienti che ricevevano rispettivamente tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e un inibitore del TNF, al mese 12. Al mese 24, l'innalzamento è risultato rispettivamente del 12,71%, 18,14% e 3,64%.
- Il colesterolo HDL medio è aumentato dell'11,71%, 13,63% e 2,82% nei pazienti che ricevevano rispettivamente tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e un inibitore del TNF, al mese 12. Al mese 24, l'innalzamento è risultato rispettivamente dell'11,58%, 13,54% e 1,42%.
Negli studi clinici sulla CU, le variazioni nei lipidi osservate con il trattamento con tofacitinib erano simili alle variazioni osservate negli studi clinici sull'AR.
Infarto del miocardio
Artrite reumatoide
In un ampio studio (N=4362) randomizzato di sicurezza, successivo all'autorizzazione, condotto in pazienti con artrite reumatoide di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, i tassi di incidenza (95% IC) di infarto miocardico non fatale per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF sono stati rispettivamente di 0,37 (0,22-0,57), 0,33 (0,19-0,53) e 0,16 (0,07-0,31) pazienti con eventi per 100 pazienti-anno. Sono stati segnalati pochi casi di infarto miocardico fatale con tassi simili nei pazienti trattati con tofacitinib rispetto agli inibitori del TNF (vedere paragrafi 4.4 e 5.1). Lo studio richiedeva almeno 1500 pazienti da seguire per 3 anni.
Tumori maligni, escluso NMSC
Artrite reumatoide
In un ampio studio (N=4362) randomizzato di sicurezza, successivo all'autorizzazione, condotto in pazienti con artrite reumatoide di età pari o superiore a 50 anni con almeno un fattore di rischio cardiovascolare aggiuntivo, i tassi di incidenza (95% IC) di cancro del polmone per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF sono stati rispettivamente di 0,23 (0,12-0,40), 0,32 (0,18-0,51) e 0,13 (0,05-0,26) pazienti con eventi per 100 pazienti-anno (vedere paragrafi 4.4 e 5.1). Lo studio richiedeva almeno 1500 pazienti da seguire per 3 anni.
I tassi di incidenza (IC al 95%) di linfoma per tofacitinib 5 mg due volte al giorno, tofacitinib 10 mg due volte al giorno e inibitori del TNF sono stati rispettivamente di 0,07 (0,02-0,18), 0,11 (0,04-0,24) e 0,02 (0,00-0,10) pazienti con eventi per 100 pazienti-anno (vedere paragrafi 4.4 e 5.1).
Popolazione pediatrica
Artrite idiopatica giovanile poliarticolare e PsA giovanile
Le reazioni avverse nei pazienti con JIA nel programma di sviluppo clinico erano coerenti per tipo e frequenza con quelle osservate in pazienti adulti con AR, ad eccezione di alcune infezioni (influenza, faringite, sinusite, infezione virale) e disordini gastrointestinali o generici (dolore addominale, nausea, vomito, piressia, cefalea, tosse), che sono stati più comuni nella popolazione pediatrica con JIA. Il MTX è stato il csDMARD utilizzato in concomitanza con maggiore frequenza (il Giorno 1, 156 dei 157 pazienti in trattamento con csDMARD hanno assunto MTX). Non esistono dati sufficienti riguardo al profilo di sicurezza di tofacitinib usato in concomitanza con qualsiasi altro csDMARD.
Infezioni
Nella fase in doppio cieco dello studio registrativo di Fase 3 (Studio JIA-I), l'infezione è stata la reazione avversa più comunemente riportata (44,3%). Le infezioni erano generalmente di severità da lieve a moderata.
Nella popolazione dell'analisi integrata di sicurezza, 7 pazienti hanno avuto infezioni gravi durante il trattamento con tofacitinib nel periodo di riferimento (fino a 28 giorni dopo l'ultima dose del farmaco in studio), con un tasso di incidenza di 1,92 pazienti con eventi per 100 pazienti-anno: polmonite, empiema epidurale (con sinusite e ascesso subperiosteo), cisti pilonidale, appendicite, pielonefrite da escherichia, ascesso di un arto e IVU.
Nella popolazione dell'analisi integrata di sicurezza , 3 pazienti hanno avuto eventi non gravi di herpes zoster all'interno della finestra di segnalazione che rappresenta un tasso di incidenza di 0,82 pazienti con eventi per 100 pazienti-anno. Un (1) altro paziente ha avuto un evento di HZ grave al di fuori della finestra di segnalazione.
Eventi epatici
Per essere idonei all'arruolamento, i pazienti nello studio registrativo sulla JIA dovevano presentare livelli di AST e ALT inferiori a 1,5 volte il limite superiore della norma. Nella popolazione dell'analisi integrata di sicurezza , 2 pazienti hanno presentato aumenti di ALT ≥ 3 volte l'ULN in occasione di 2 visite consecutive. Nessuno dei due eventi ha soddisfatto i criteri della legge di Hy. Entrambi i pazienti erano in terapia di base con MTX e ciascun evento si è risolto dopo la sospensione di MTX e la sospensione permanente di tofacitinib.
Test di laboratorio
Le variazioni nei test di laboratorio dei pazienti con JIA nel programma di sviluppo clinico erano coerenti con quelli osservati nei pazienti adulti con AR. I pazienti nello studio registrativo sulla JIA dovevano presentare una conta piastrinica ≥ 100.000 cellule/mm3 per essere idonei all'arruolamento, pertanto non sono disponibili informazioni per i pazienti con JIA con conta piastrinica < 100.000 cellule/mm3 prima di iniziare il trattamento con tofacitinib.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l'autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite l'Agenzia Italiana del Farmaco Sito web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Sovradosaggio
Cosa fare se avete preso una dose eccessiva di Xeljanz
In caso di sovradosaggio, si raccomanda di monitorare il paziente per segni e sintomi di reazioni avverse. Non esiste un antidoto specifico per il sovradosaggio con Tofacitinib. Il trattamento deve essere sintomatico e di supporto.
I dati farmacocinetici, fino ad una singola dose di 100 mg, in volontari sani indicano che oltre il 95% della dose somministrata dovrebbe essere eliminata entro 24 ore.
Scadenza
4 anni.
Conservazione
Questo medicinale non richiede alcuna temperatura particolare di conservazione.
Conservare nella confezione originale per proteggere il medicinale dall'umidità.
Foglietto Illustrativo
Fonti Ufficiali
Servizi Avanzati



© 2022 EDRA S.p.A. - P.iva 08056040960
DPO - dpo@lswr.it