
Valganciclovir Aurobindo
Ultimo aggiornamento: 24/07/2023
Cos'è Valganciclovir Aurobindo?
Valganciclovir Aurobindo è un farmaco a base del principio attivo
Valganciclovir Cloridrato, appartenente alla categoria degli
Antiretrovirali e nello specifico
Nucleosidi e nucleotidi, esclusi gli inibitori della transcrittasi inversa. E' commercializzato in Italia dall'azienda
Aurobindo Pharma (Italia) S.r.l..
Valganciclovir Aurobindo può essere prescritto con Ricetta RR - medicinali soggetti a prescrizione medica.
Valganciclovir Aurobindo può essere prescritto con Ricetta RR - medicinali soggetti a prescrizione medica.
Confezioni
Valganciclovir Aurobindo 450 mg 60 compresse rivestite con film
Informazioni commerciali sulla prescrizione
Titolare: Aurobindo Pharma (Italia) S.r.l.
Ricetta: RR - medicinali soggetti a prescrizione medica
Classe: A
Principio attivo: Valganciclovir Cloridrato
Gruppo terapeutico: Antiretrovirali
ATC: J05AB14 - Valganciclovir
Forma farmaceutica: compresse rivestite
Ricetta: RR - medicinali soggetti a prescrizione medica
Classe: A
Principio attivo: Valganciclovir Cloridrato
Gruppo terapeutico: Antiretrovirali
ATC: J05AB14 - Valganciclovir
Forma farmaceutica: compresse rivestite
Se sei un professionista, potrai trovare le schede tecniche complete e molto altro nell'area riservata di Codifa.it
Indicazioni
Perché si usa Valganciclovir Aurobindo? A cosa serve?
Valganciclovir Aurobindo è indicato per il trattamento di induzione e mantenimento della retinite da citomegalovirus (CMV) in adulti con sindrome da immunodeficienza acquisita (AIDS).
Valganciclovir Aurobindo è indicato per la prevenzione della malattia da CMV in adulti e bambini (dalla nascita ai 18 anni di età) CMV-negativi che hanno ricevuto un trapianto di organo solido da un donatore CMV-positivo.
Posologia
Come usare Valganciclovir Aurobindo: Posologia
Posologia
Attenzione - Per evitare il sovradosaggio è fondamentale attenersi rigorosamente alle raccomandazioni posologiche (vedere paragrafi 4.4 e 4.9).
Dopo somministrazione orale, valganciclovir è rapidamente e largamente metabolizzato a ganciclovir. Una dose di valganciclovir 900 mg due volte al giorno (bid) per via orale è terapeuticamente equivalente a una dose di ganciclovir 5 mg/kg bid per via endovenosa.
Trattamento della retinite da citomegalovirus (CMV)
Pazienti adulti
Trattamento di induzione della retinite da CMV:
Nei pazienti con retinite da CMV attiva, la dose raccomandata è di 900 mg di valganciclovir (due compresse di Valganciclovir Aurobindo 450 mg) due volte/die per 21 giorni, da assumere ove possibile a stomaco pieno. Un trattamento di induzione prolungato può aumentare il rischio di tossicità midollare (vedere paragrafo 4.4).
Trattamento di mantenimento della retinite da CMV:
Dopo il trattamento di induzione, o nei pazienti con retinite da CMV inattiva, la dose raccomandata è di 900 mg di valganciclovir (due compresse di Valganciclovir Aurobindo 450 mg) una volta/die, da assumere ove possibile a stomaco pieno. I pazienti in cui la retinite va incontro a peggioramento possono ripetere il trattamento di induzione; è tuttavia necessario considerare la possibilità di una resistenza virale al farmaco.
La durata del trattamento di mantenimento deve essere stabilita su base individuale.
Popolazione pediatrica
La sicurezza e l'efficacia di valganciclovir nel trattamento della retinite da CMV in pazienti pediatrici non sono state determinate nell'ambito di studi clinici adeguati e ben controllati.
Prevenzione della malattia da CMV in caso di trapianto di organo solido:
Pazienti adulti
Per i pazienti sottoposti a trapianto di rene, la dose raccomandata è di 900 mg (due compresse di Valganciclovir Aurobindo 450 mg) una volta/die, da iniziare entro 10 giorni dal trapianto e proseguire per i 100 giorni successivi al medesimo. È possibile continuare la profilassi fino a 200 giorni dopo il trapianto (vedere paragrafi 4.4, 4.8 e 5.1).
Per i pazienti sottoposti a trapianto di organi solidi diversi dal rene, la dose raccomandata è di 900 mg (due compresse di Valganciclovir Aurobindo 450 mg) una volta/die, da iniziare entro 10 giorni dal trapianto e proseguire per i 100 giorni successivi al medesimo.
Ove possibile, le compresse devono essere assunte a stomaco pieno.
Popolazione pediatrica
In pazienti pediatrici, a partire dalla nascita, sottoposti a trapianto di organo solido che sono a rischio di sviluppare la malattia da CMV, la dose raccomandata di valganciclovir in singola somministrazione giornaliera si basa sulla superficie corporea (BSA) e sulla clearance della creatinina (ClCr) derivata dalla formula di Schwartz (ClCrS), e si calcola usando l'equazione sotto riportata:
Dose pediatrica (mg) = 7 x BSA x ClCrS (vedere la formula di Mosteller per la BSA e la formula di Schwartz per la clearance della creatinina riportate sotto).
Se la clearance della creatinina calcolata con la formula di Schwartz supera i 150 ml/min/1,73 m2, nell'equazione si dovrà utilizzare un valore massimo di 150 ml/min/1,73 m2:
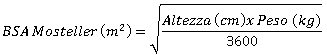
|
Clearance della creatinina di Schwartz (ml/min/1,73 m2) =
|
k x Altezza (cm)
Creatinina sierica (mg/dL)
|
|
I valori di k forniti si basano sul metodo di Jaffe per la misurazione della creatinina sierica e possono richiedere una correzione quando si utilizzano metodi enzimatici.
* Per appropriate sottopopolazioni può essere richiesta anche una riduzione del valore di k (ad esempio in pazienti pediatrici con basso peso alla nascita).
Per i pazienti pediatrici sottoposti a trapianto di rene, la dose raccomandata in mg (7 x BSA x ClCrS) una volta al giorno deve iniziare entro i 10 giorni successivi al trapianto e deve proseguire fino a 200 giorni dopo il trapianto.
Per i pazienti pediatrici sottoposti a un trapianto di organo solido diverso dal rene, la dose raccomandata in mg (7 x BSA x ClCrS) una volta al giorno deve iniziare entro i 10 giorni successivi al trapianto e deve proseguire fino a 100 giorni dopo il trapianto.
Tutte le dosi calcolate devono essere arrotondate al più vicino incremento da 25 mg per la dose effettivamente erogabile. Se la dose calcolata supera i 900 mg, si dovrà somministrare una dose massima di 900 mg. La soluzione orale è la formulazione da preferire poiché consente di somministrare una dose calcolata in base alla formula sopra riportata; è tuttavia possibile utilizzare le compresse rivestite con film di valganciclovir se le dosi calcolate risultano comprese in un intervallo pari al 10% delle dosi disponibili per le compresse e il paziente è in grado di ingerire le compresse. Ad esempio, se la dose calcolata è compresa tra 405 mg e 495 mg, si potrà assumere una compressa da 450 mg.
Si raccomanda di monitorare regolarmente i livelli di creatinina sierica e valutare le modifiche di altezza e peso corporeo e adattare la dose in modo appropriato durante il periodo di trattamento profilattico.
Istruzioni particolari di dosaggio
Popolazione pediatrica:
Il dosaggio nei pazienti pediatrici sottoposti a trapianto di organo solido deve essere personalizzato sulla base della funzionalità renale e della superficie corporea del soggetto.
Pazienti anziani:
La sicurezza e l'efficacia non sono state determinate in questa popolazione di pazienti e non sono stati condotti studi in soggetti adulti di età superiore ai 65 anni. Poiché l'avanzamento dell'età comporta una riduzione della clearance renale, valganciclovir deve essere somministrato in pazienti anziani tenendo sotto stretto controllo lo stato dei reni (vedere tabella sottostante e paragrafo 5.2).
Pazienti con danno renale:
I livelli di creatinina sierica o la clearance della creatinina stimata devono essere monitorati con attenzione. Sono richiesti aggiustamenti della dose in base alla clearance della creatinina, come indicato nella tabella sotto riportata (vedere paragrafi 4.4 e 5.2).
La clearance della creatinina (mL/min) può essere stimata in base alla creatinina sierica, applicando la seguente formula:
| Sesso maschile = |
(140-età [anni]) x (peso corporeo [kg]) (140-età [anni]) x (peso corporeo [kg]) |
| Sesso femminile = | 0,85 x il valore maschile |
|
ClCr (ml/min)
|
Dose di induzione di valganciclovir
|
Dose di mantenimento/prevenzione di valganciclovir
|
|
≥ 60
|
900 mg (2 compresse) due volte/die
|
900 mg (2 compresse) due volte/die
|
|
40 – 59
|
450 mg (1 compressa) due volte/die
|
450 mg (1 compressa) una volta/die
|
|
25 – 39
|
450 mg (1 compressa) una volta/die
|
450 mg (1 compressa) ogni 2 giorni
|
|
10 – 24
|
450 mg (1 compressa) ogni 2 giorni
|
450 mg (1 compressa) due volte a settimana
|
|
< 10
|
non raccomandata
|
non raccomandata
|
Pazienti sottoposti a emodialisi:
Per i pazienti emodializzati (ClCr <10 mL/min) non è possibile fornire raccomandazioni riguardanti la dose. Valganciclovir non deve pertanto essere utilizzato in questi pazienti (vedere paragrafi 4.4 e 5.2).
Pazienti con compromissione epatica:
La sicurezza e l'efficacia di valganciclovir compresse nei pazienti con compromissione epatica non sono state studiate (vedere paragrafo 5.2).
Pazienti con leucopenia, neutropenia, anemia, trombocitopenia e pancitopenia gravi:
Prima di iniziare la terapia, vedere il paragrafo 4.4.
In caso di peggioramento significativo delle conte ematiche durante la terapia con valganciclovir, si dovrà prendere in considerazione un trattamento con fattori di crescita emopoietici e/o l'interruzione della somministrazione (vedere paragrafo 4.4).
Modo di somministrazione
Valganciclovir è somministrato per via orale e, ove possibile, deve essere assunto a stomaco pieno (vedere paragrafo 5.2).
Per i pazienti pediatrici che non sono in grado di deglutire le compresse rivestite con film di valganciclovir, può essere somministrato valganciclovir polvere per soluzione orale.
Precauzioni che devono essere prese prima della manipolazione o della somministrazione del medicinale
Le compresse non devono essere rotte o frantumate. Poiché valganciclovir è considerato un potenziale teratogeno e cancerogeno nell'uomo, si deve prestare attenzione durante la manipolazione di compresse rotte (vedere paragrafo 4.4). Evitare il contatto diretto di compresse rotte o frantumate con cute o mucose. In caso di contatto, lavare accuratamente con acqua e sapone; sciacquare accuratamente gli occhi con acqua sterile o con acqua normale, se l'acqua sterile non fosse disponibile.
Controindicazioni
Quando non dev'essere usato Valganciclovir Aurobindo
Valganciclovir è controindicato in pazienti con ipersensibilità a valganciclovir, ganciclovir o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
Valganciclovir è controindicato durante l'allattamento (vedere paragrafo 4.6).
Avvertenze speciali e precauzioni di impiego
Cosa serve sapere prima di prendere Valganciclovir Aurobindo
Ipersensibilità crociata
Data la stretta somiglianza di struttura chimica tra ganciclovir, aciclovir e penciclovir, è possibile una reazione di ipersensibilità crociata con questi farmaci. Si deve pertanto esercitare cautela quando si prescrive valganciclovir a pazienti con ipersensibilità nota ad aciclovir o penciclovir (o ai relativi profarmaci, rispettivamente valaciclovir e famciclovir).
Mutagenicità, teratogenicità, carcinogenicità, fertilità e contraccezione
Prima di avviare il trattamento con valganciclovir, i pazienti devono essere informati dei potenziali rischi per il feto. In studi con ganciclovir, condotti sull'animale, sono stati riscontrati gli effetti mutageni, teratogeni, aspermatogeni, cancerogeni e soppressori della fertilità femminile. Valganciclovir va pertanto considerato un agente potenzialmente teratogeno e cancerogeno nell'uomo, che può provocare difetti di nascita e tumori (vedere paragrafo 5.3). Si ritiene inoltre probabile che valganciclovir provochi un'inibizione temporanea o permanente della spermatogenesi. Si deve indicare alle donne potenzialmente fertili di fare uso di un contraccettivo efficace durante il trattamento e per almeno 30 giorni dopo il trattamento. Si deve indicare agli uomini di utilizzare metodi contraccettivi di barriera durante il trattamento e per almeno i 90 giorni successivi, salvo nel caso in cui il rischio di gravidanza per la partner sia escluso con certezza (vedere paragrafi 4.6, 4.8 e 5.3).
Nel lungo termine, valganciclovir può provocare cancerogenicità e tossicità riproduttiva.
Mielosoppressione
Nei pazienti trattati con valganciclovir (e ganciclovir) sono state osservate gravi leucopenia, neutropenia, anemia, trombocitopenia, pancitopenia, insufficienza midollare e anemia aplastica gravi. La terapia non deve essere avviata se la conta assoluta dei neutrofili è inferiore a 500 cellule/μL, se la conta piastrinica è inferiore a 25.000/μL, oppure se il livello di emoglobina è inferiore a 8 g/dL (vedere paragrafi 4.2 e 4.8).
Nei casi in cui la profilassi si protragga oltre i 100 giorni, si deve tenere in considerazione il possibile rischio per il paziente di sviluppare leucopenia e neutropenia (vedere paragrafi 4.2, 4.8 e 5.1).
Valganciclovir deve essere usato con cautela nei pazienti con preesistente citopenia o anamnesi positiva per citopenia farmaco-correlata, e nei pazienti sottoposti a radioterapia.
Durante la terapia si raccomanda di effettuare un monitoraggio della conta completa delle cellule ematiche e delle piastrine. Nei pazienti con danno renale ed in quelli pediatrici deve essere garantita un più frequente monitoraggio ematologico, quantomeno ogni volta che il paziente si reca presso il centro trapianti. Nei pazienti che sviluppano leucopenia, neutropenia, anemia e/o trombocitopenia gravi si raccomanda di prendere in considerazione il trattamento con fattori di crescita emopoietici e/o l'interruzione della somministrazione (vedere paragrafi 4.2).
Differenza della biodisponibilità con ganciclovir per via orale
La biodisponibilità di ganciclovir dopo una dose singola da 900 mg di valganciclovir è pari a circa il 60%, rispetto a circa il 6% dopo somministrazione di 1.000 mg di ganciclovir (in capsule) per via orale. Un'eccessiva esposizione a ganciclovir può essere associata a reazioni avverse potenzialmente letali. Si consiglia pertanto di attenersi scrupolosamente alle raccomandazioni riguardanti la dose quando si istituisce la terapia, si passa dalla terapia di induzione a quella di mantenimento e nei pazienti che possono passare da ganciclovir per via orale a valganciclovir in quanto valganciclovir non può sostituire in modo biunivoco ganciclovir in capsule. I pazienti che sostituiscono la terapia con ganciclovir in capsule devono essere informati del rischio di sovradosaggio nel caso in cui dovessero assumere un numero di compresse di valganciclovir superiore a quanto prescritto (vedere paragrafi 4.2 e 4.9).
Danno renale
Nei pazienti con compromissione della funzione renale, sono necessari aggiustamenti della dose in base alla clearance della creatinina (vedere paragrafi 4.2 e 5.2).
Le compresse rivestite con film di valganciclovir non devono essere utilizzate in pazienti emodializzati (vedere paragrafi 4.2 e 5.2).
Uso con altri medicinali
In pazienti che assumono imipenem-cilastatina e ganciclovir sono state riferite convulsioni. Valganciclovir non deve essere utilizzato in concomitanza con imipenem-cilastatina salvo nel caso in cui i potenziali benefici superino i potenziali rischi (vedere paragrafo 4.5).
I pazienti trattati con valganciclovir e (a) didanosina, (b) farmaci che inducono mielosoppressione (ad es. zidovudina), o (c) sostanze che esercitano effetti sulla funzione renale devono essere sottoposti a un attento monitoraggio per rilevare eventuali segni di ulteriore tossicità (vedere paragrafo 4.5).
Lo studio clinico controllato in cui è stato utilizzato valganciclovir per la profilassi della malattia da CMV nel trapianto non comprendeva pazienti sottoposti a trapianto di polmone e intestino, come indicato nel paragrafo 5.1; l'esperienza nei pazienti sottoposti a questi tipi di trapianto è pertanto limitata.
Interazioni con altri medicinali e altre forme di interazione
Quali farmaci o alimenti possono modificare l'effetto di Valganciclovir Aurobindo
Interazioni farmacologiche con valganciclovir
Non sono stati eseguiti studi di interazione in vivo con valganciclovir. Poiché valganciclovir è rapidamente e largamente metabolizzato a ganciclovir; le interazioni associate a valganciclovir sono le stesse che ci si aspetta per ganciclovir.
Interazioni farmacologiche con ganciclovir
Interazioni farmacocinetiche
Probenecid
La somministrazione di probenecid con ganciclovir per via orale ha determinato una riduzione statisticamente significativa della clearance renale di ganciclovir (20%) con conseguente aumento statisticamente significativo dell'esposizione (40%). Queste variazioni sono risultate in linea con un meccanismo di interazione che prevede una competizione per la secrezione tubulare renale. I pazienti che assumono probenecid e valganciclovir devono pertanto essere sottoposti a un attento monitoraggio per rilevare un'eventuale tossicità da ganciclovir.
Didanosina
Le concentrazioni plasmatiche di didanosina sono risultate costantemente aumentate quando questo farmaco è stato somministrato con ganciclovir per via endovenosa. A dosi di 5 e 10 mg/kg/die di ganciclovir somministrato per via endovenosa, è stato osservato un aumento dell'AUC di didanosina tra il 38 e il 67 %, a conferma di un'interazione farmacocinetica durante la somministrazione concomitante di questi due farmaci. Non è stato osservato un effetto significativo sulle concentrazioni di ganciclovir. I pazienti devono essere strettamente controllati per l'eventuale comparsa di tossicità da didanosina, come la pancreatite (vedere paragrafo 4.4).
Altri antiretrovirali
Gli isoenzimi del citocromo P450 non influiscono sulla farmacocinetica di ganciclovir. Pertanto non si prevedono interazioni farmacocinetiche con inibitori della proteasi e inibitori della trascrittasi inversa non nucleosidici.
Interazioni farmacodinamiche
Imipenem-cilastatina
Sono state riportate convulsioni in pazienti che assumevano contemporaneamente ganciclovir e imipenem-cilastatina e la possibilità di un'interazione farmacodinamica tra questi due farmaci non può essere esclusa.
Questi farmaci non devono essere utilizzati contemporaneamente a meno che i potenziali benefici non superino i potenziali rischi (vedere paragrafo 4.4).
Zidovudina
Sia zidovudina che ganciclovir possono causare neutropenia e anemia. Quando somministrati contemporaneamente, può verificarsi un'interazione farmacodinamica. Alcuni pazienti possono non tollerare la somministrazione concomitante di entrambi i farmaci a dosaggio pieno (vedere paragrafo 4.4).
Possibili interazioni farmacologiche
La tossicità può risultare incrementata quando ganciclovir/valganciclovir è somministrato in concomitanza con altri farmaci notoriamente mielosoppressivi o associati a danno renale, quali analoghi nucleosidici (ad es. zidovudina, didanosina, stavudina) e nucleotidici (ad es. tenofovir, adefovir), immunosoppressori (ad es. ciclosporina, tacrolimus, micofenolato mofetile), agenti antineoplastici (ad es. doxorubicina, vinblastina, vincristina, idrossiurea) e agenti anti-infettivi (trimetoprim/sulfonamidi, dapsone, amfotericina B, flucitosina, pentamidina). Pertanto, l'impiego concomitante di questi farmaci con valganciclovir deve essere valutato soltanto nel caso in cui i potenziali benefici superino i potenziali rischi (vedere paragrafo 4.4).
Fertilità, gravidanza e allattamento
Contraccezione in uomini e donne
Come risultato del rischio potenziale di tossicità riproduttiva e teratogenicità, le donne in età fertile devono essere informate sulla necessità di utilizzare metodi contraccettivi efficaci durante il trattamento con ganciclovir e per almeno 30 giorni dopo la sua conclusione. Si deve indicare ai pazienti di sesso maschile di utilizzare metodi contraccettivi di barriera durante il trattamento con valganciclovir e per almeno i 90 giorni successivi, salvo nel caso in cui il rischio di gravidanza per la partner sia escluso con certezza (vedere paragrafi 4.4 e 5.3).
Gravidanza
La sicurezza dell'uso di valganciclovir in gravidanza non è stata stabilita. Il suo metabolita attivo, ganciclovir, si diffonde facilmente attraverso la placenta umana. In base al meccanismo farmacologico d'azione e alla tossicità riproduttiva osservata in studi sull'animale con ganciclovir (vedere paragrafo 5.3) esiste un rischio teorico di teratogenicità nell'uomo.
Valganciclovir in compresse non deve essere usato in gravidanza, salvo nel caso in cui il beneficio terapeutico per la madre superi il rischio potenziale di teratogenicità per il feto.
Allattamento
Benché non sia noto se ganciclovir sia escreto nel latte materno umano, non si può escludere la possibilità che ciò avvenga e provochi reazioni avverse gravi nel lattante. I dati emersi dagli studi sugli animali indicano che ganciclovir è escreto nel latte di femmine di ratto in fase di allattamento.
Perciò durante il trattamento con valganciclovir l'allattamento al seno deve essere interrotto (vedere paragrafo 4.3 e 5.3).
Fertilità
Non sono disponibili dati sugli effetti di valganciclovir sulla fertilità nell'uomo. Gli studi di fertilità non sono stati ripetuti su valganciclovir a causa della sua rapida ed estesa conversione a ganciclovir nell'organismo. Negli studi sull'animale ganciclovir è associato ad una compromissione della fertilità (vedere paragrafo 5.3).
Effetti sulla capacità di guidare veicoli e sull'uso di macchinari
Non sono stati condotti studi per valutare gli effetti sulla capacità di guidare veicoli e sull'uso di macchinari.
Con l'uso di valganciclovir e/o ganciclovir sono state riferite convulsioni, capogiro e confusione. Tali effetti, se presenti, possono influenzare l'esecuzione di attività che richiedono attenzione, tra cui la capacità del paziente di guidare veicoli e usare macchinari.
Effetti indesiderati
Quali sono gli effetti collaterali di Valganciclovir Aurobindo
a. Riassunto del profilo di sicurezza
Valganciclovir è un profarmaco di ganciclovir che viene metabolizzato rapidamente ed abbondantemente a ganciclovir dopo somministrazione orale. Gli effetti indesiderati noti associati all'uso di ganciclovir possono essere attesi anche con valganciclovir. Tutte le reazioni avverse al farmaco osservate nel corso degli studi con valganciclovir sono state precedentemente osservate con ganciclovir. Pertanto, le reazioni avverse al farmaco segnalate con l'impiego di ganciclovir per via endovenosa od orale (formulazione non più disponibile) o con valganciclovir sono incluse nella relativa tabella delle reazioni avverse al farmaco di seguito riportata.
Nei pazienti trattati con valganciclovir/ganciclovir le reazioni avverse al farmaco più gravi e comuni sono state quelle ematologiche che includono neutropenia, anemia e trombocitopenia (vedere paragrafo 4.4).
Le categorie di frequenza riportate nella tabella delle reazioni avverse provengono da una popolazione aggregata di pazienti (n=1704) in terapia di mantenimento con ganciclovir o valganciclovir. Fanno eccezione reazione anafilattica, agranulocitosi e granulocitopenia, le cui categorie di frequenza derivano dall'esperienza successiva all'immissione in commercio del prodotto. Le reazioni avverse sono elencate utilizzando la classificazione per organi e classi secondo MedDRA. Le categorie di frequenza sono definite in base alla seguente convenzione: molto comune (≥ 1/10), comune (≥ 1/100, < 1/10), non comune (≥ 1/1.000, < 1/100), raro (≥ 1/10.000, < 1/1.000) e molto raro (< 1/10.000).
Il profilo di sicurezza complessivo di ganciclovir/valganciclovir è coerente con quello segnalato nelle popolazioni affette da HIV o sottoposte a trapianto, ad eccezione del distacco della retina, riportato soltanto in pazienti con retinite da CMV. Nella frequenza di alcune reazioni si riscontrano tuttavia delle differenze.
Valganciclovir è associato a un rischio maggiore di diarrea rispetto a ganciclovir per via endovenosa. Piressia, infezioni da candida, depressione, neutropenia severa (ANC <500/μl) e reazioni cutanee sono segnalate più comunemente nei pazienti affetti da HIV. Nei pazienti sottoposti a trapianto, invece, sono riportate con maggiore frequenza disfunzione renale ed epatica.
b. Elenco tabulare delle reazioni avverse al farmaco
|
Reazione avversa al farmaco (ADR)
(MedDRA)
Classificazione per organi e sistemi
|
Categoria di frequenza
|
|
Infezioni e infestazioni:
|
|
|
Infezioni da candida, compresa candidiasi orale
|
Molto comune
|
|
Infezione delle vie respiratorie superiori
|
|
|
Sepsi
|
Comune
|
|
Influenza
|
|
|
Infezioni del tratto urinario
|
|
|
Cellulite
|
|
|
Patologie del sistema emolinfopoietico:
|
|
|
Neutropenia
|
Molto comune
|
|
Anemia
|
|
|
Trombocitopenia
|
Comune
|
|
Leucopenia
|
|
|
Pancitopenia
|
|
|
Insufficienza midollare
|
Non comune
|
|
Anemia aplastica
|
Raro
|
|
Agranulocitosi*
|
|
|
Granulocitopenia*
|
|
|
Disturbi del sistema immunitario:
|
|
|
Ipersensibilità
|
Comune
|
|
Reazione anafilattica*
|
Raro
|
|
Disturbi del metabolismo e della nutrizione:
|
|
|
Diminuzione dell'appetito
|
Molto comune
|
|
Perdita di peso
|
Comune
|
|
Disturbi psichiatrici:
|
|
|
Depressione
|
Comune
|
|
Stato confusionale
|
|
|
Ansia
|
|
|
Agitazione
|
Non comune
|
|
Disturbi psicotici
|
|
|
Pensiero anormale
|
|
|
Allucinazioni
|
|
|
Patologie del sistema nervoso:
|
|
|
Cefalea
|
Molto comune
|
|
Insonnia
|
Comune
|
|
Neuropatia periferica
|
|
|
Capogiro
|
|
|
Parestesia
|
|
|
Ipoestesia
|
|
|
Convulsioni
|
|
|
Disgeusia (alterazione del gusto)
|
|
|
Tremore
|
Non comune
|
|
Patologie dell'occhio:
|
|
|
Compromissione della vista
|
Comune
|
|
Distacco di retina**
|
|
|
Miodesopsie (mosche volanti)
|
|
|
Dolore oculare
|
|
|
Congiuntivite
|
|
|
Edema maculare
|
|
|
Patologie dell'orecchio e del labirinto:
|
|
|
Dolore all'orecchio
|
Comune
|
|
Sordità
|
Non comune
|
|
Patologie cardiache:
|
|
|
Aritmie
|
Non comune
|
|
Patologie vascolari:
|
|
|
Ipotensione
|
Comune
|
|
Patologie respiratorie, toraciche e mediastiniche:
|
|
|
Tosse
|
Molto comune
|
|
Dispnea
|
|
|
Patologie gastrointestinali:
|
|
|
Diarrea
|
Molto comune
|
|
Nausea
|
|
|
Vomito
|
|
|
Dolore addominale
|
|
|
Dispepsia
|
Comune
|
|
Flatulenza
|
|
|
Dolore addominale superiore
|
|
|
Stipsi
|
|
|
Ulcerazione della bocca
|
|
|
Disfagia
|
|
|
Distensione addominale
|
|
|
Pancreatite
|
|
|
Patologie epatobiliari:
|
|
|
Aumento dei livelli di fosfatasi alcalina ematica
|
Comune
|
|
Alterazione della funzionalità epatica
|
|
|
Aumento dei livelli di aspartato aminotransferasi
|
|
|
Aumento dei livelli di alanina aminotransferasi
|
|
|
Patologie della cute e del tessuto sottocutaneo:
|
|
|
Dermatite
|
Molto comune
|
|
Sudorazione notturna
|
Comune
|
|
Prurito
|
|
|
Eruzione cutanea
|
|
|
Alopecia
|
|
|
Secchezza cutanea
|
Non comune
|
|
Orticaria
|
|
|
Patologie del sistema muscoloscheletrico e del tessuto connettivo:
|
|
|
Dolore alla schiena
|
Comune
|
|
Mialgia
|
|
|
Artralgia
|
|
|
Spasmi muscolari
|
|
|
Patologie renali e urinarie:
|
|
|
Danno renale
|
Comune
|
|
Riduzione della clearance renale della creatinina
|
|
|
Aumento dei livelli di creatinina ematica
|
|
|
Insufficienza renale
|
Non comune
|
|
Ematuria
|
|
|
Patologie dell'apparato riproduttivo e della mammella:
|
|
|
Infertilità maschile
|
Non comune
|
|
Patologie sistemiche e condizioni relative alla sede di somministrazione:
|
|
|
Piressia
|
Molto comune
|
|
Affaticamento
|
|
|
Dolore
|
Comune
|
|
Brividi
|
|
|
Malessere
|
|
|
Astenia
|
|
|
Dolore toracico
|
Non comune
|
* Le categorie di frequenza di queste reazioni avverse provengono dall'esperienza successiva all'immissione in commercio del prodotto.
** Il distacco di retina è stato segnalato soltanto in pazienti con AIDS trattati per retinite da CMV.
Descrizione di una selezione di reazioni avverse
Neutropenia
Non è possibile prevedere il rischio di neutropenia in base al numero dei neutrofili prima del trattamento. Solitamente la neutropenia insorge durante la prima o la seconda settimana del trattamento di induzione. Di norma, la conta cellulare rientra nei valori normali 2-5 giorni dopo la sospensione del farmaco o la riduzione della dose (vedere paragrafo 4.4).
Trombocitopenia
I pazienti con basse conte piastriniche (< 100.000/μl) al basale, presentano un aumento del rischio di sviluppare trombocitopenia. I pazienti con immunosoppressione iatrogena, a causa del trattamento con farmaci immunosoppressori, sono esposti a un rischio più elevato di sviluppare trombocitopenia rispetto ai pazienti affetti da AIDS (vedere paragrafo 4.4). La trombocitopenia severa può essere associata a un sanguinamento potenzialmente letale.
Influenza della durata del trattamento o dell'indicazione terapeutica sulle reazioni avverse
L'insorgenza di neutropenia severa (ANC < 500/μl) si osserva con maggiore frequenza nei pazienti con retinite da CMV (14%) in terapia con valganciclovir e ganciclovir per via endovenosa od orale rispetto ai pazienti sottoposti a trapianto di organo solido in terapia con valganciclovir o ganciclovir per via orale. Nei pazienti trattati con valganciclovir o ganciclovir orale fino a 100 giorni dopo il trapianto, l'incidenza di neutropenia severa è risultata pari, rispettivamente, al 5% e al 3%, mentre nei pazienti trattati con valganciclovir fino a 200 giorni dopo il trapianto l'incidenza di neutropenia severa è risultata pari al 10%.
Rispetto ai soggetti con retinite da CMV, nei pazienti sottoposti a trapianto di organo solido trattati fino a 100 o 200 giorni dopo il trapianto con valganciclovir e ganciclovir orale, è stato riscontrato un aumento maggiore dei livelli di creatinina sierica. Un'alterata funzionalità renale, tuttavia, rappresenta una caratteristica comune nei pazienti sottoposti a trapianto di organo solido.
Nei pazienti sottoposti a trapianto di rene ad alto rischio il profilo di sicurezza complessivo di valganciclovir è rimasto invariato con l'estensione della profilassi fino a 200 giorni. È stata riportata leucopenia con un'incidenza leggermente maggiore nel braccio trattato fino a 200 giorni, mentre l'incidenza di neutropenia, anemia e trombocitopenia è risultata simile in entrambi i bracci.
c. Popolazione pediatrica
Valganciclovir è stato studiato in 179 pazienti pediatrici sottoposti a trapianto di organo solido che sono a rischio di sviluppare malattia da CMV (di età compresa tra le 3 settimane e i 16 anni) e in 133 neonati con malattia da CMV congenita sintomatica (di età compresa tra i 2 e i 31 giorni), con una durata di esposizione al ganciclovir compresa tra i 2 e i 200 giorni.
Le reazioni avverse segnalate con maggiore frequenza durante il trattamento negli studi clinici pediatrici sono state diarrea, nausea, neutropenia, leucopenia e anemia.
In soggetti sottoposti a trapianto di organo solido, il profilo di sicurezza complessivo nei pazienti pediatrici è risultato simile a quello degli adulti. È stata segnalata neutropenia con incidenza lievemente superiore nei due studi condotti su pazienti pediatrici sottoposti a trapianto di organo solido rispetto agli adulti, ma non è emersa una correlazione tra neutropenia ed eventi avversi infettivi nella popolazione pediatrica.
In pazienti pediatrici sottoposti a trapianto di rene, il prolungamento dell'esposizione al valganciclovir fino a 200 giorni non è stato associato ad un aumento complessivo dell'incidenza di eventi avversi. L'incidenza di neutropenia grave (ANC < 500/μl) è risultata superiore nei pazienti pediatrici sottoposti a trapianto di rene trattati fino a 200 giorni rispetto ai pazienti pediatrici trattati fino a 100 giorni e rispetto a pazienti adulti sottoposti a trapianto di rene trattati fino a 100 o fino a 200 giorni (vedere paragrafo 4.4).
Sono disponibili soltanto dati limitati relativi a neonati o lattanti con infezione da CMV congenita sintomatica trattati con valganciclovir; tuttavia, la sicurezza sembra in linea con il profilo di sicurezza noto di valganciclovir/ganciclovir.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l'autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite il sistema nazionale di segnalazione all'indirizzo http://www.agenziafarmaco.gov.it/content/come-segnalare-una-sospetta-reazione-avversa.
Sovradosaggio
Cosa fare se avete preso una dose eccessiva di Valganciclovir Aurobindo
Esperienze di sovradosaggio con valganciclovir e ganciclovir per via endovenosa
Si prevede che un sovradosaggio di valganciclovir possa essere associato a un aumento della tossicità renale (vedere paragrafi 4.2 e 4.4).
Nel corso di studi clinici e dell'esperienza post-marketing sono stati riferiti sovradosaggi di ganciclovir per via endovenosa, alcuni con esito fatale. In alcuni di questi casi non sono stati riferiti eventi avversi; la maggior parte dei pazienti ha manifestato uno o più dei seguenti eventi avversi:
- Tossicità ematologica: mielosoppressione inclusa pancitopenia, aplasia midollare, leucopenia, neutropenia, granulocitopenia.
- Epatotossicità: epatite, disturbi della funzionalità epatica.
- Tossicità renale: peggioramento dell'ematuria in un paziente con preesistente danno renale, insufficienza renale acuta, livelli elevati di creatinina.
- Tossicità gastrointestinale: dolore addominale, diarrea, vomito.
- Neurotossicità: tremore generalizzato, convulsioni.
Nei pazienti a cui viene somministrata una dose eccessiva di valganciclovir il ricorso all'emodialisi e all'idratazione può essere utile per ridurre i livelli plasmatici circolanti del farmaco (vedere paragrafo 5.2).
Scadenza
3 anni.
Conservazione
Questo medicinale non richiede alcuna condizione particolare di conservazione.
Foglietto Illustrativo
Fonti Ufficiali
Servizi Avanzati



© 2022 EDRA S.p.A. - P.iva 08056040960
DPO - dpo@lswr.it