
Lutathera
Ultimo aggiornamento: 17/04/2024
Cos'è Lutathera?
Lutathera è un farmaco a base del principio attivo
Lutezio-177lu-oxodotreotide, appartenente alla categoria degli
Radiofarmaceutici terapeutici e nello specifico
Radiofarmaceutici terapeutici vari. E' commercializzato in Italia dall'azienda
Novartis Farma S.p.A..
Lutathera può essere prescritto con Ricetta OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile.
Lutathera può essere prescritto con Ricetta OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile.
Confezioni
Lutathera 370 MBq/mL soluzione per infusione, uso endovenoso 25 ml 1 flaconcino
Informazioni commerciali sulla prescrizione
Titolare: Advanced Accelerator Applications SA
Concessionario: Novartis Farma S.p.A.
Ricetta: OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile
Classe: H
Principio attivo: Lutezio-177lu-oxodotreotide
Gruppo terapeutico: Radiofarmaceutici terapeutici
ATC: V10XX04 - Lutezio (177Lu) oxodotreotide
Forma farmaceutica: soluzione
Concessionario: Novartis Farma S.p.A.
Ricetta: OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile
Classe: H
Principio attivo: Lutezio-177lu-oxodotreotide
Gruppo terapeutico: Radiofarmaceutici terapeutici
ATC: V10XX04 - Lutezio (177Lu) oxodotreotide
Forma farmaceutica: soluzione
Se sei un professionista, potrai trovare le schede tecniche complete e molto altro nell'area riservata di Codifa.it
Indicazioni
Perché si usa Lutathera? A cosa serve?
Lutathera è indicato in pazienti adulti per il trattamento di tumori neuroendocrini gastroenteropancreatici (NET-GEP) ben differenziati (G1 e G2), progressivi, non asportabili o metastatici, positivi ai recettori per la somatostatina,.
Posologia
Come usare Lutathera: Posologia
Importanti istruzioni di sicurezza
La somministrazione di Lutathera deve essere condotta esclusivamente da persone autorizzate a manipolare radiofarmaci in strutture cliniche appositamente designate (vedere paragrafo 6.6) e solo dopo aver sottoposto il paziente all'esame di un medico qualificato.
Identificazione del paziente
Prima di iniziare il trattamento con Lutathera, l'imaging recettoriale della somatostatina (scintigrafia o tomografia ad emissione di positroni [PET]) deve confermare la sovraespressione di tali recettori nel tessuto tumorale con una captazione da parte del tumore pari almeno alla normale captazione epatica.
Posologia
Adulti
Il regime di trattamento raccomandato con Lutathera negli adulti consiste in 4 infusioni da 7.400 MBq ciascuna. L'intervallo consigliato tra una somministrazione e la successiva è di 8 settimane (±1 settimana).
Nelle rispettive sezioni di seguito sono fornite informazioni sulle modifiche della dose per la gestione di reazioni avverse al farmaco severe o non tollerabili.
Soluzione di amminoacidi
Come forma di protezione renale, è necessario somministrare per 4 ore una soluzione di amminoacidi, contenente L-lisina e L-arginina, per via endovenosa (vedere la composizione nelle Tabelle 1 e 2). L'infusione della soluzione di amminoacidi deve iniziare 30 minuti prima dell'avvio dell'infusione di Lutathera. L'infusione della soluzione di amminoacidi e di Lutathera attraverso un accesso venoso separato in ciascun braccio del paziente è il metodo preferito. Tuttavia, se non è possibile utilizzare due linee endovenose a causa di un accesso venoso insufficiente o di una preferenza istituzionale/clinica, la soluzione di amminoacidi e di Lutathera possono essere infuse attraverso la stessa linea mediante una valvola a tre vie, tenendo in considerazione la portata e il mantenimento dell'accesso venoso. La dose della soluzione di amminoacidi non deve essere ridotta anche se viene somministrata una dose ridotta di Lutathera.
Una soluzione di amminoacidi contenente solo L-lisina e L-arginina nelle quantità specificate nella Tabella 1 è considerata il medicinale di scelta, per il minore volume totale da infondere e la più bassa osmolalità.
La soluzione di amminoacidi può essere preparata come prodotto estemporaneo, conformemente alle buone pratiche di preparazione dell'ospedale per i prodotti medicinali sterili e secondo la formulazione specificata in Tabella 1.
Tabella 1 Composizione della soluzione estemporanea di amminoacidi
|
Composto
|
Quantità
|
|
L-lisina cloridrato
|
25 g*
|
|
L-arginina cloridrato
|
25 g**
|
|
Soluzione iniettabile di sodio cloruro 9 mg/mL (0,9%), o acqua per preparazioni iniettabili
|
1 L
|
|
*equivalente a 20,0 g di L-lisina
**equivalente a 20,7 g di L-arginina
|
|
In alternativa, se conformi alle specifiche descritte in Tabella 2, possono essere utilizzate soluzioni di amminoacidi disponibili in commercio.
Tabella 2 Specifiche delle soluzioni di amminoacidi disponibili in commercio
|
Caratteristica
|
Specifica
|
|
L-lisina cloridrato
|
tra 18 e 25 g*
|
|
L-arginina cloridrato
|
tra 18 e 25 g**
|
|
Volume
|
da 1 a 2 L
|
|
Osmolalità
|
<1 200 mOsmol/kg
|
|
*equivalente a 14,4-20 g di L-lisina
**equivalente a 14,9-20,7 g di L-arginina
|
|
Monitoraggio del trattamento
Prima di ogni somministrazione e durante il trattamento con Lutathera, è necessario effettuare test di laboratorio per valutare le condizioni del paziente e, ove necessario, adattare il protocollo terapeutico (dose, intervallo di infusione, numero di infusioni) (vedere Tabella 3).
I test di laboratorio minimi necessari prima di ogni infusione sono:
- Ematologia (emoglobina [Hb], conta leucocitaria con conta differenziale, conta piastrinica)
- Funzionalità renale (creatinina e clearance della creatinina secondo la formula di Cockcroft-Gault)
- Funzionalità epatica (alanina aminotransferasi [ALT], aspartato aminotransferasi [AST], albumina sierica, rapporto internazionale normalizzato (INR) e bilirubina)
Questi test di laboratorio devono essere eseguiti almeno una volta nelle 2-4 settimane che precedono la somministrazione, e poco prima della somministrazione. Si raccomanda inoltre di eseguire questi test ogni 4 settimane per almeno 3 mesi dall'ultima infusione di Lutathera e successivamente ogni 6 mesi per poter rilevare eventuali reazioni avverse ritardate (vedere paragrafo 4.8). Potrebbe essere necessario modificare il dosaggio in base ai risultati dei test (vedere Tabella 3).
Modifica della dose
La gestione di reazioni avverse al farmaco severe o non tollerate può richiedere una sospensione temporanea del trattamento (estensione dell'intervallo di somministrazione da 8 a 16 settimane), una riduzione della dose o l'interruzione permanente del trattamento con Lutathera (vedere Tabella 3 e Figura 1).
Tabella 3 Modifiche della dose di Lutathera raccomandate in caso di reazioni avverse al farmaco
|
Reazione avversa al farmaco
|
Severità della reazione avversa al farmaco
|
Modifica della dose
|
|
Trombocitopenia
|
Prima comparsa di:
Grado 2 (piastrine <75–50 x 109/L)
Grado 3 (piastrine <50–25 x 109/L)
Grado 4 (piastrine <25 x 109/L)
|
Sospendere il trattamento fino a completa o parziale risoluzione (Grado da 0 a 1).
Riprendere Lutathera a 3 700 MBq (100 mCi) in pazienti con risoluzione parziale o completa. Se la riduzione della dose non dà luogo ad una trombocitopenia di Grado 2, 3 o 4, somministrare 7 400 MBq (200 mCi) di Lutathera come dose successiva.
Interrompere definitivamente Lutathera in caso di trombocitopenia di Grado 2 o superiore che richieda un intervallo tra le somministrazioni oltre le 16 settimane.
|
|
Grado 2, 3 o 4 ricorrente
|
Interrompere definitivamente Lutathera
|
|
|
Anemia e neutropenia
|
Prima comparsa di anemia:
Grado 3 (Hb <8,0 g/dL); trasfusione indicata
Grado 4 (sequele pericolose per la vita)
Prima comparsa di neutropenia:
Grado 3 (conta assoluta dei neutrofili [ANC] <1,0–0,5 x 109/L)
Grado 4 (ANC <0,5 x 109/L)
|
Sospendere il trattamento fino a completa o parziale risoluzione (Grado 0, 1, o 2).
Riprendere Lutathera a 3 700 MBq (100 mCi) in pazienti con risoluzione parziale o completa. Se la riduzione della dose non dà luogo ad anemia o neutropenia di Grado 3 o 4, somministrare 7 400 MBq (200 mCi) come dose successiva.
Interrompere definitivamente Lutathera in caso di anemia o neutropenia di Grado 3 o superiore che richieda un intervallo tra le somministrazioni oltre le 16 settimane.
|
|
Grado 3 o 4 ricorrente
|
Interrompere definitivamente Lutathera
|
|
|
Tossicità renale
|
Prima comparsa di:
· Clearance della creatinina inferiore a 40 mL/min; calcolata mediante la formula di Cockcroft-Gault utilizzando il peso corporeo attuale, o
· Aumento del 40% della creatinina sierica rispetto al basale, o
· Diminuzione del 40% della clearance della creatinina rispetto al basale, calcolata mediante la formula di Cockcroft-Gault utilizzando il peso corporeo attuale
|
Sospendere il trattamento fino a risoluzione o ritorno al basale.
Riprendere Lutathera a 3 700 MBq (100 mCi) in pazienti con risoluzione o ritorno al basale. Se la riduzione della dose non dà luogo a tossicità renale, somministrare 7 400 MBq (200 mCi) di Lutathera come dose successiva.
Interrompere definitivamente Lutathera in caso di tossicità renale che richieda un intervallo tra le somministrazioni oltre le 16 settimane.
|
|
Tossicità renale ricorrente
|
Interrompere definitivamente Lutathera
|
|
|
Epatotossicità
|
Prima comparsa di:
· Bilirubinemia 3 volte superiore al limite superiore di norma (Grado 3 o 4), o
· Albuminemia inferiore a 30 g/L con INR >1,5
|
Sospendere il trattamento fino a risoluzione o ritorno al basale.
Riprendere Lutathera a 3 700 MBq (100 mCi) in pazienti con risoluzione o ritorno al basale. Se la riduzione della dose non dà luogo ad epatotossicità, somministrare 7 400 MBq (200 mCi) di Lutathera come dose successiva.
Interrompere definitivamente Lutathera in caso di epatotossicità che richieda un intervallo tra le somministrazioni oltre le 16 settimane.
|
|
Epatotossicità ricorrente
|
Interrompere definitivamente Lutathera
|
|
|
Qualsiasi altra reazione avversa al farmaco1 di Grado 3 o 4 secondo CTCAE*
|
Prima comparsa di Grado 3 o 4
|
Sospendere il trattamento fino a completa o parziale risoluzione.
Riprendere Lutathera a 3 700 MBq (100 mCi) in pazienti con risoluzione completa o parziale. Se la riduzione della dose non dà luogo tossicità di Grado 3 o 4, somministrare 7 400 MBq (200 mCi) di Lutathera come dose successiva.
Interrompere definitivamente Lutathera in caso di reazione avversa al farmaco di Grado 3 o 4 che richieda un intervallo tra le somministrazioni oltre le 16 settimane.
|
|
Grado 3 o 4 ricorrente
|
Interrompere definitivamente Lutathera
|
|
|
1 Non è richiesta alcuna modifica della dose per le tossicità ematologiche di Grado 3 o di Grado 4 dovute esclusivamente a linfopenia.
* CTCAE: Common Terminology Criteria for Adverse Events, National Cancer Institute
|
||
Figura 1 Riepilogo delle istruzioni per modificare la dose
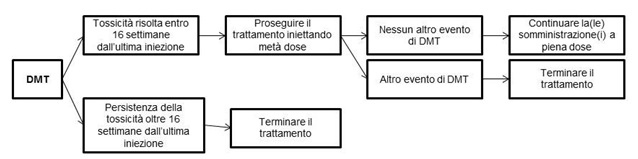
DMT: tossicità modificante la dose
Altri fattori per considerare l'interruzione temporanea del trattamento con Lutathera includono la comparsa di una malattia intercorrente (ad es. infezione delle vie urinarie) che il medico ritiene possa aumentare i rischi associati alla somministrazione di Lutathera e che deve essere risolta o stabilizzata al fine di riprendere il trattamento, o un intervento chirurgico importante nel qual caso il trattamento deve essere sospeso per 12 settimane dopo la data dell'intervento chirurgico.
Popolazioni speciali
Anziani
Non è richiesta alcuna modifica della dose nei pazienti di età pari o superiore a 65 anni poiché l'esperienza clinica non ha individuato differenze di risposta tra pazienti anziani e pazienti più giovani. Tuttavia, dal momento che è stato descritto un aumento del rischio di ematotossicità nei pazienti anziani (≥70 anni), è consigliabile un follow-up ravvicinato che consenta di adattare rapidamente la dose (DMT) in questa popolazione.
Compromissione renale
Poiché nei pazienti con compromissione renale è possibile un aumento dell'esposizione alle radiazioni, è richiesta un'attenta valutazione dell'attività da somministrare. Il profilo farmacocinetico e la sicurezza di lutezio (177Lu) oxodotreotide in pazienti con compromissione renale severa al basale (clearance della creatinina <30 mL/min secondo la formula di Cockcroft-Gault) o con malattia renale allo stadio terminale non sono stati studiati. Il trattamento con Lutathera è controindicato in pazienti con insufficienza renale con clearance della creatinina <30 mL/min (vedere paragrafo 4.3). Il trattamento con Lutathera non è raccomandato nei pazienti con clearance della creatinina <40 mL/min al basale (utilizzando la formula di Cockcroft-Gault). Non è raccomandato alcun aggiustamento della dose nei pazienti con compromissione renale con clearance della creatinina ≥40 mL/min al basale. Tuttavia, poiché questo medicinale è noto per essere sostanzialmente escreto per via renale, si raccomanda una maggior frequenza nel monitoraggio della funzionalità renale in questi pazienti poiché sono maggiormente a rischio di tossicità.
Per maggiori dettagli circa il trattamento di pazienti con tossicità renale, vedere la Tabella 3 nel paragrafo 4.2 e il paragrafo 4.4.
Compromissione epatica
Poiché nei pazienti con compromissione epatica è possibile un aumento dell'esposizione alle radiazioni, è richiesta un'attenta valutazione dell'attività da somministrare. Il profilo farmacocinetico e la sicurezza di lutezio (177Lu) oxodotreotide nei pazienti con compromissione epatica severa al basale (bilirubina totale >3 volte il limite superiore di norma, indipendentemente dal livello di AST) non sono stati studiati. I pazienti con compromissione epatica al basale con bilirubina totale >3 volte rispetto al limite superiore del range di normalità o albuminemia <30 g/L e INR >1,5 devono essere trattati con Lutathera solo dopo attenta valutazione del beneficio-rischio. Non è raccomandata alcuna modifica della dose nei pazienti con compromissione epatica lieve o moderata al basale.
Per ulteriori dettagli sul trattamento di pazienti con epatotossicità, vedere la Tabella 3 nel paragrafo 4.2 e il paragrafo 4.4.
Popolazione pediatrica
Non esiste alcuna indicazione per un uso specifico di Lutathera nella popolazione pediatrica per l'indicazione di trattamento di NET-GEP (eccetto per neuroblastoma, neuroganglioblastoma e feocromocitoma).
Modo di somministrazione
Lutathera è per uso endovenoso. È un radiofarmaco pronto all'uso, solo per uso singolo.
Istruzioni per la somministrazione
Per la somministrazione della dose raccomandata possono essere utilizzati il metodo per gravità, il metodo con pompa peristaltica o il metodo con pompa a siringa. Gli operatori sanitari che eseguono il trattamento possono utilizzare altri metodi ritenuti appropriati e sicuri, specialmente quando è richiesta una riduzione della dose.
Quando si utilizza il metodo per gravità o il metodo con pompa peristaltica, Lutathera deve essere infuso direttamente dal suo contenitore originale. Il metodo con pompa peristaltica o il metodo con pompa a siringa devono essere utilizzati quando si somministra una dose ridotta di Lutathera a seguito di una modifica della dose a causa di una reazione avversa (vedere la Tabella 3 nel paragrafo 4.2). L'uso del metodo per gravità per somministrare una dose ridotta di Lutathera può comportare la somministrazione di un volume errato di Lutathera se la dose non viene adattata prima della somministrazione. Devono essere adottate misure precauzionali di radioprotezione indipendentemente dal metodo di somministrazione utilizzato (vedere paragrafo 6.6).
La seguente tabella riassume l'intera procedura di somministrazione di Lutathera:
Tabella 4 Procedura per la somministrazione dell'antiemetico, della soluzione di amminoacidi e di Lutathera
|
Agente somministrato
|
Ora di inizio
(min.)
|
Velocità di infusione
(mL/h)
|
Durata
|
|
Antiemetico
|
Almeno 30 minuti prima della soluzione di amminoacidi
|
Come da riassunto delle caratteristiche del prodotto
|
Come da riassunto delle caratteristiche del prodotto
|
|
Soluzione di amminoacidi, o preparata estemporaneamente (1 L) o commerciale (da 1 a 2 L)
|
0
|
250–500 a seconda del volume
|
4 ore
|
|
Lutathera con soluzione iniettabile di sodio cloruro 9 mg/mL (0,9%)
|
30
|
Fino a 400
|
30 ± 10 minuti
|
Per le istruzioni sul metodo di preparazione e sui metodi di somministrazione endovenosa, vedere il paragrafo 12.
Per le raccomandazioni in caso di stravaso, vedere il paragrafo 4.4.
Controindicazioni
Quando non dev'essere usato Lutathera
- Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
- Gravidanza accertata o sospetta o quando la gravidanza non è stata esclusa (vedere paragrafo 4.6).
- Insufficienza renale con clearance della creatinina <30 mL/min.
Avvertenze speciali e precauzioni di impiego
Cosa serve sapere prima di prendere Lutathera
Giustificazione del rapporto beneficio-rischio individuale
Per ogni paziente, l'esposizione alle radiazioni deve essere giustificata in rapporto al probabile beneficio. L'attività somministrata deve essere, in ogni caso, quella ragionevolmente più bassa possibile in grado di ottenere l'effetto terapeutico richiesto.
Considerato il meccanismo di azione e il profilo di tollerabilità di Lutathera, si raccomanda di non iniziare il trattamento con Lutathera in pazienti negativi al recettore della somatostatina o con lesioni viscerali miste in base all'imaging del recettore della somatostatina.
Mielosoppressione
A causa dei potenziali effetti ematologici indesiderati, la conta ematica deve essere monitorata al basale e prima di ogni dose di Lutathera durante il trattamento e fino alla risoluzione di eventuali tossicità (vedere paragrafo 4.2). I pazienti con compromissione midollare e i pazienti che hanno ricevuto una precedente chemioterapia o radioterapia a fasci esterni (che abbia interessato più del 25% del midollo osseo) possono essere a maggiore rischio di tossicità ematologica durante il trattamento con Lutathera. Il trattamento non è raccomandato nei pazienti con compromissione severa della funzione ematologica al basale e durante il trattamento (ad es. Hb <4,9 mmol/L o 8 g/dL, piastrine <75 x 109/L, o leucociti <2 x 109/L) a meno che non sia dovuta esclusivamente a linfopenia.
Sindrome mielodisplastica e leucemia acuta
Dopo il trattamento con Lutathera sono state osservate sindrome mielodisplastica (MDS) ad esordio tardivo e leucemia acuta (AL) (vedere paragrafo 4.8) verificatesi rispettivamente dopo circa 29 mesi (9-45) e dopo circa 55 mesi (32-125) dalla prima infusione di Lutathera. L'eziologia di queste neoplasie mieloidi secondarie correlate alla terapia (t-MNs) non è chiara. Sono stati ipotizzati come possibili fattori di rischio e/o fattori predittivi di MDS/AL un'età >70 anni, la compromissione della funzionalità renale, la citopenia al basale, il numero di precedenti terapie, la pregressa esposizione ad agenti chemioterapici (specificamente agenti alchilanti) e le precedenti radioterapie.
Tossicità renale
Poiché lutezio (177Lu) oxodotreotide viene quasi esclusivamente eliminato attraverso il sistema renale, è obbligatorio somministrare contemporaneamente una soluzione di amminoacidi contenente gli amminoacidi L-lisina e L-arginina. La soluzione di amminoacidi contribuirà a ridurre il riassorbimento del lutezio (177Lu) oxodotreotide attraverso i tubuli prossimali, con conseguente riduzione significativa della dose assorbita a livello renale (vedere paragrafo 4.2). Quando l'infusione concomitante della soluzione di amminoacidi raccomandata viene somministrata per un periodo di 4 ore, è stata riportata una riduzione media dell'esposizione renale alla radiazione di circa il 47%.
I pazienti devono essere incoraggiati a rimanere idratati e a urinare frequentemente prima, il giorno stesso e il giorno dopo la somministrazione di Lutathera (ad es. 1 bicchiere d'acqua ogni ora).
La funzionalità renale, determinata dalla creatinina sierica e dalla clearance della creatinina calcolata utilizzando la formula di Cockcroft-Gault, deve essere valutata al basale, durante e per almeno il primo anno dopo il trattamento (vedere paragrafo 4.2).
I pazienti con compromissione renale al basale, o con anomalie renali o del tratto urinario possono essere a maggior rischio di tossicità a causa dell'aumentata esposizione alle radiazioni (vedere paragrafo 4.2).
Per i pazienti con clearance della creatinina <50 mL/min deve inoltre essere considerato un aumento del rischio di iperkaliemia transitoria dovuto alla somministrazione della soluzione di amminoacidi (vedere Avvertenze e precauzioni specifiche per la somministrazione concomitante di una soluzione di amminoacidi nefroprotettiva).
Epatotossicità
Poiché molti pazienti avviati alla terapia con Lutathera presentano metastasi epatiche, può essere comune osservare pazienti con alterata funzionalità epatica al basale. I pazienti con metastasi epatiche o pre-esistente compromissione epatica in stadio avanzato possono essere maggiormente a rischio di epatotossicità a causa dell'esposizione alle radiazioni. Pertanto, durante il trattamento si consiglia di monitorare ALT, AST, bilirubina, albumina sierica e INR (vedere paragrafo 4.2).
Ipersensibilità
Casi di reazioni di ipersensibilità (inclusi eventi isolati di angioedema) sono stati segnalati in pazienti trattati con Lutathera nella fase post-marketing (vedere paragrafo 4.8). In caso di gravi reazioni di ipersensibilità, l'infusione di Lutathera in corso deve essere interrotta immediatamente. Medicinali e attrezzature appropriati per gestire tali reazioni devono essere disponibili per uso immediato.
Nausea e vomito
Per prevenire la nausea e il vomito dovuti al trattamento, un bolo endovenoso di un antiemetico deve essere iniettato almeno 30 minuti prima di avviare l'infusione della soluzione di amminoacidi per raggiungere la piena efficacia antiemetica (vedere paragrafo 4.2).
Uso concomitante di analoghi della somatostatina
La somatostatina e i suoi analoghi si legano in modo competitivo ai recettori della somatostatina e possono interferire con l'efficacia di Lutathera (vedere paragrafo 4.5).
Crisi ormonali neuroendocrine
Poiché possono verificarsi crisi dovute a eccessivo rilascio di ormoni o di sostanze bioattive dopo il trattamento con Lutathera, in alcuni casi deve essere considerato il monitoraggio attraverso il ricovero notturno dei pazienti (ad es., pazienti con scarso controllo farmacologico dei sintomi). In caso di crisi ormonali, i trattamenti raccomandati sono: somministrazione endovenosa di analoghi della somatostatina a dosi elevate, di liquidi per via endovenosa, di corticosteroidi, e correzione dei disturbi elettrolitici nei pazienti con diarrea e/o vomito.
Sindrome da lisi tumorale
Dopo la terapia con medicinali contenenti lutezio-177 sono stati segnalati casi di sindrome da lisi tumorale. I pazienti con una storia di insufficienza renale e un carico tumorale elevato presentano un rischio più elevato e devono essere trattati con maggiore cautela. La funzionalità renale e l'equilibrio elettrolitico devono essere valutati al basale e durante il trattamento.
Regole di radioprotezione
I pazienti trattati con Lutathera devono essere tenuti lontani da altre persone durante la somministrazione e fino al raggiungimento dei limiti di emissione delle radiazioni previsti dalle leggi applicabili, solitamente entro 4-5 ore dopo la somministrazione del medicinale. L'operatore sanitario deve stabilire quando il paziente può lasciare l'area controllata dell'ospedale, vale a dire il momento in cui l'esposizione alle radiazioni di terze parti non supera le soglie regolamentari.
I pazienti devono essere incoraggiati a rimanere idratati e a urinare frequentemente prima, il giorno stesso ed il giorno dopo la somministrazione di Lutathera (ad es. 1 bicchiere d'acqua ogni ora) per facilitare l'eliminazione del medicinale. I pazienti devono essere incoraggiati anche a defecare ogni giorno e, ove necessario, ad aiutarsi con un lassativo. L'urina e le feci devono essere smaltite conformemente alle normative nazionali.
A meno che non vi sia contaminazione cutanea del paziente dovuta, ad esempio, a fuoriuscite dal sistema di infusione o a incontinenza urinaria, non ci si attendono contaminazioni di radioattività della cute o della massa di materiale vomitato. Tuttavia, durante le normali cure mediche o gli esami con dispositivi medici o altri strumenti vengono a contatto con la cute (ad es., elettrocardiogramma [ECG]), si consiglia di osservare misure di protezione di base, come indossare guanti, applicare materiali/elettrodi prima di avviare l'infusione del radiofarmaco e sostituirli una volta terminate le misurazioni, e, infine, monitorare il livello di radioattività dell'apparecchiatura dopo l'uso.
Prima di essere dimesso, il paziente deve essere istruito sulle regole di radioprotezione necessarie a cui attenersi durante le interazioni con altri membri della stessa famiglia e con altre persone in generale e sulle precauzioni generali che il paziente deve adottare durante le attività quotidiane dopo il trattamento (come indicato nel prossimo paragrafo e nel foglio illustrativo) per ridurre al minimo l'esposizione alle radiazioni di altre persone.
Dopo ogni somministrazione, possono essere prese in considerazione le seguenti raccomandazioni generali insieme alle procedure e ai regolamenti nazionali, locali e istituzionali:
- Deve essere limitata la stretta vicinanza (meno di 1 metro) ad altre persone per 7 giorni.
- Nel caso di bambini e/o donne in gravidanza, la stretta vicinanza (meno di 1 metro) deve essere limitata a meno di 15 minuti al giorno per 7 giorni.
- I pazienti devono dormire in una camera da letto separata dalle altre persone per 7 giorni.
- I pazienti devono dormire in una camera da letto separata dai bambini e/o dalle donne in gravidanza per 15 giorni.
Misure consigliate in caso di stravaso
Si devono indossare guanti impermeabili monouso. L'infusione del medicinale deve essere interrotta immediatamente ed il dispositivo di somministrazione (catetere, ecc.) rimosso. Informare il medico specialista in medicina nucleare e il radiofarmacista.
Tutti i materiali del dispositivo di somministrazione devono essere conservati per potere misurare la radioattività residua, l'attività effettivamente somministrata, e deve essere determinata la dose assorbita. L'area di stravaso deve essere delimitata con una penna indelebile e, se possibile, se ne deve fare una foto. Inoltre, si raccomanda di registrare il tempo e il volume stimato dello stravaso.
Per continuare l'infusione di Lutathera, è obbligatorio utilizzare un nuovo catetere, eventualmente inserendolo in un accesso venoso controlaterale.
Nessun medicinale supplementare può essere somministrato omolateralmente alla sede dello stravaso.
Al fine di accelerare la dispersione del medicinale e prevenire la sua stagnazione nei tessuti, si raccomanda di aumentare il flusso sanguigno sollevando il braccio interessato. A seconda del caso, deve essere presa in considerazione l'aspirazione del liquido di stravaso, un'iniezione di lavaggio con soluzione iniettabile di sodio cloruro 9 mg/mL (0,9%), o l'applicazione di compresse calde o di un impacco caldo al sito di infusione per accelerare la vasodilatazione.
Si devono trattare eventuali sintomi, in particolare l'infiammazione e/o il dolore. A seconda della situazione, il medico specialista in medicina nucleare deve informare il paziente circa i rischi connessi alle lesioni da stravaso e fornire suggerimenti circa i possibili trattamenti e le necessarie esigenze di follow-up. L'area di stravaso deve essere controllata fino alla dimissione del paziente dall'ospedale. A seconda della severità dello stravaso, l'evento deve essere dichiarato come reazione avversa.
Pazienti con incontinenza urinaria
Onde evitare la diffusione di contaminazione radioattiva, durante i primi 2 giorni successivi alla somministrazione di questo medicinale, si devono adottare precauzioni particolari nei pazienti con incontinenza urinaria. Ciò include la manipolazione di qualsiasi materiale eventualmente contaminato da urina.
Pazienti con metastasi cerebrali
Poiché non sono disponibili dati di efficacia per i pazienti con metastasi cerebrali note, in questi pazienti deve essere valutato caso per caso il rapporto beneficio-rischio.
Neoplasie secondarie maligne
L'esposizione a radiazioni ionizzanti è collegata all'induzione del cancro e al potenziale sviluppo di difetti ereditari. La dose di radiazioni risultante dall'esposizione terapeutica può determinare una maggiore incidenza di cancro e di mutazioni geniche. In tutti i casi è necessario garantire che i rischi dovuti all'esposizione alle radiazioni siano inferiori a quelli derivati dalla malattia stessa.
Altri pazienti con fattori di rischio
I pazienti con una delle seguenti condizioni sono più inclini a sviluppare reazioni avverse. Pertanto, si raccomanda una maggior frequenza nel monitoraggio di questi pazienti durante il trattamento. In caso di tossicità modificante la dose, fare riferimento alla Tabella 3.
- Metastasi ossee;
- Precedenti radioterapie metaboliche oncologiche con composti con (131I) o qualsiasi altra terapia che utilizza sorgenti radioattive non schermate;
- Storia di altre neoplasie, a meno che il paziente non sia considerato in remissione da almeno 5 anni.
Contraccezione nei maschi e nelle femmine
Alle pazienti di sesso femminile in età fertile deve essere consigliato di usare un metodo contraccettivo efficace durante il trattamento e per 7 mesi dopo l'ultima dose di Lutathera (vedere paragrafo 4.6).
Ai pazienti di sesso maschile con partner femminili in età fertile deve essere consigliato di usare un metodo contraccettivo efficace durante il trattamento e per 4 mesi dopo l'ultima dose di Lutathera (vedere paragrafo 4.6).
Avvertenze e precauzioni specifiche per la somministrazione concomitante di una soluzione di amminoacidi nefroprotettiva
Iperkaliemia
Nei pazienti in trattamento con arginina e lisina può verificarsi un aumento transitorio dei livelli sierici di potassio, che solitamente rientrano nell'intervallo di normalità entro 24 ore dall'inizio dell'infusione della soluzione di amminoacidi. I pazienti con ridotta clearance della creatinina possono essere maggiormente a rischio di iperkaliemia transitoria (vedere “Tossicità renale” nel paragrafo 4.4).
I livelli sierici di potassio devono essere valutati prima di ogni somministrazione di una soluzione di amminoacidi. In caso di iperkaliemia, verificare l'anamnesi del paziente per iperkaliemia e le terapie concomitanti. L'iperkaliemia deve conseguentemente essere corretta prima di iniziare l'infusione.
In caso di pre-esistente iperkaliemia clinicamente significativa, un secondo monitoraggio deve confermare che l'iperkaliemia è stata corretta con successo, prima dell'infusione di una soluzione di amminoacidi. Il paziente deve essere strettamente monitorato per segni e sintomi di iperkaliemia, ad es. dispnea, debolezza, intorpidimento, dolore toracico e manifestazioni cardiache (anomalie di conduzione e aritmie cardiache). Deve essere eseguito un elettrocardiogramma (ECG) prima di dimettere il paziente.
Durante l'infusione devono essere monitorati i parametri vitali indipendentemente dai livelli sierici basali di potassio. I pazienti devono essere incoraggiati a rimanere idratati e a urinare frequentemente prima, il giorno stesso e il giorno dopo la somministrazione (ad es. 1 bicchiere d'acqua ogni ora) per facilitare l'eliminazione del potassio sierico in eccesso.
Nel caso in cui i sintomi di iperkaliemia si sviluppino durante l'infusione della soluzione di amminoacidi, devono essere adottate le opportune misure correttive. In caso di iperkaliemia severa sintomatica, deve essere considerata la sospensione dell'infusione della soluzione di amminoacidi, considerando il rapporto beneficio-rischio tra la protezione renale e l'iperkaliemia acuta.
Insufficienza cardiaca
A causa delle potenziali complicazioni cliniche legate al sovraccarico di liquidi, si deve prestare cautela nell'uso di arginina e lisina in pazienti con insufficienza cardiaca severa definita di grado III o IV secondo la classificazione della NYHA (New York Heart Association). I pazienti con insufficienza cardiaca severa definita di grado III o IV secondo la classificazione della NYHA devono essere trattati solo dopo un'attenta valutazione del benefico-rischio, tenendo conto del volume e dell'osmolalità della soluzione di amminoacidi.
Acidosi metabolica
È stata osservata acidosi metabolica con l'utilizzo di soluzioni di amminoacidi complesse somministrate come parte dei protocolli di nutrizione parenterale totale (TPN). Le modifiche dell'equilibrio acido-base alterano l'equilibrio del potassio extra-intracellulare e un rapido aumento del potassio nel plasma può essere associato allo sviluppo di acidosi.
Avvertenze speciali
Contenuto di sodio
Questo medicinale contiene fino a 3,5 mmol (81,1 mg) di sodio per flaconcino, equivalente al 4% dell'assunzione massima giornaliera raccomandata dall'OMS che corrisponde a 2 g di sodio per un adulto.
Per le precauzioni relative al rischio ambientale vedere paragrafo 6.6.
Interazioni con altri medicinali e altre forme di interazione
Quali farmaci o alimenti possono modificare l'effetto di Lutathera
Analoghi della somatostatina
La somatostatina ed i suoi analoghi si legano competitivamente ai recettori della somatostatina e possono interferire con l'efficacia di Lutathera. Pertanto, nei 30 giorni precedenti alla somministrazione di questo medicinale, deve essere evitata la somministrazione di analoghi della somatostatina a lunga durata d'azione. Se necessario, i pazienti possono essere trattati con analoghi della somatostatina a breve durata d'azione fino a 24 ore prima della somministrazione di Lutathera.
Glucocorticoidi
Sussistono evidenze a favore della capacità dei glucocorticoidi di indurre sottoregolazione dei recettori di sottotipo 2 della somatostatina (SSTR2). Pertanto, come misura cautelativa, devono essere evitate ripetute somministrazioni di alte dosi di glucocorticoidi durante il trattamento con Lutathera. Nei pazienti con storia di uso cronico di glucocorticoidi si deve valutare attentamente la presenza di una sufficiente espressione del recettore della somatostatina. Non è noto se l'uso intermittente dei glucocorticoidi per la prevenzione di nausea e vomito durante la somministrazione di Lutathera possa indurre una sottoregolazione degli SSTR2. Come misura cautelativa, anche l'uso di glucocorticoidi come trattamento antiemetico preventivo deve essere evitato. Nel caso in cui il trattamento somministrato per la prevenzione di nausea e vomito prima dell'infusione della soluzione di amminoacidi risulti insufficiente, può essere utilizzata una singola dose di glucocorticoidi, a condizione che la somministrazione non avvenga prima dell'inizio dell'infusione di Lutathera o entro l'ora successiva alla sua conclusione.
Fertilità, gravidanza e allattamento
Donne in età fertile
Quando è prevista la somministrazione di radiofarmaci ad una donna potenzialmente fertile, è importante determinarne l'eventuale stato di gravidanza. Qualsiasi donna che abbia saltato un ciclo mestruale deve essere considerata in stato di gravidanza fino a prova contraria. In caso di dubbio circa il potenziale stato di gravidanza (se la donna ha saltato un ciclo mestruale, se il ciclo mestruale è molto irregolare, ecc.), alla paziente devono essere proposte tecniche alternative che non utilizzino radiazioni ionizzanti (se disponibili). Prima dell'uso di Lutathera, deve essere escluso un eventuale stato di gravidanza con un test adeguato/validato.
Contraccezione nei maschi e nelle femmine
Lutathera può causare malformazioni fetali quando somministrato a donne in gravidanza.
Alle pazienti di sesso femminile in età fertile deve essere consigliato di usare un metodo contraccettivo efficace durante il trattamento e per 7 mesi dopo l'ultima dose di Lutathera.
Ai pazienti di sesso maschile con partner femminili in età fertile deve essere consigliato di usare un metodo contraccettivo efficace durante il trattamento e per 4 mesi dopo l'ultima dose di Lutathera.
Gravidanza
Non sono stati condotti studi sulla funzione riproduttiva animale con lutezio (177Lu) oxodotreotide.
Le procedure con radionuclidi eseguite su donne in stato di gravidanza comportano l'assorbimento di dosi di radiazioni anche per il feto. A causa del rischio associato alle radiazioni ionizzanti, l'uso di Lutathera è controindicato durante la gravidanza, accertata o sospetta, o quando lo stato di gravidanza non sia stato escluso (vedere paragrafo 4.3). Le donne in gravidanza devono essere avvisate dei rischi per il feto.
Allattamento
Non è noto se lutezio (177Lu) oxodotreotide venga escreto nel latte materno. Non si può escludere un rischio dovuto alle radiazioni ionizzanti per il bambino allattato. Durante il trattamento con questo medicinale deve essere evitato l'allattamento con latte materno. Se il trattamento con Lutathera durante l'allattamento con latte materno è necessario, il bambino deve essere prima svezzato.
Fertilità
Non sono stati condotti studi sugli animali finalizzati a determinare gli effetti di lutezio (177Lu) oxodotreotide sulla fertilità di entrambi i sessi. Le radiazioni ionizzanti di lutezio (177Lu) oxodotreotide possono indurre effetti tossici temporanei sulle gonadi femminili e maschili. Nel caso in cui il paziente desidera avere bambini dopo il trattamento, si raccomanda di richiedere una consultazione genetica. Prima del trattamento, è possibile parlare con i pazienti dell'opzione della crioconservazione dello sperma o degli ovociti.
Effetti sulla capacità di guidare veicoli e sull'uso di macchinari
Lutathera non altera o altera in modo trascurabile la capacità di guidare veicoli e di usare macchinari. Tuttavia, prima di porsi alla guida o di utilizzare macchinari, si devono considerare le condizioni generali del paziente ed eventuali reazioni avverse al trattamento.
Effetti indesiderati
Quali sono gli effetti collaterali di Lutathera
Riepilogo del profilo di sicurezza
Il profilo di sicurezza complessivo di Lutathera si basa sui dati raccolti nei pazienti sottoposti a studi clinici (studio NETTER-1 di fase III e studio Erasmus di fase I/II in pazienti olandesi) e da programmi di uso compassionevole.
Le reazioni avverse più comuni nei pazienti che hanno ricevuto il trattamento con Lutathera sono state nausea e vomito, insorti all'inizio dell'infusione, rispettivamente nel 58,9% e nel 45,5% dei pazienti. La causalità di nausea/vomito è confusa dall'effetto emetico della concomitante soluzione di amminoacidi somministrata per la protezione renale.
A causa della tossicità midollare di Lutathera, le reazioni avverse più attese sono state quelle correlate a tossicità ematologica: trombocitopenia (25%), linfopenia (22,3%), anemia (13,4%), pancitopenia (10,2%).
Altre reazioni avverse molto comuni riportate includono affaticamento (27,7%) e diminuzione dell'appetito (13,4%).
Al momento dell'analisi finale dello studio NETTER-1, dopo una durata mediana del follow-up di 76 mesi per ciascun braccio dello studio, il profilo di sicurezza è rimasto coerente con quello precedentemente riportato.
Elenco tabellare delle reazioni avverse
Le reazioni avverse sono elencate nella Tabella 5 in base alla frequenza e alla Classificazione per sistemi e organi (SOC - System Organ Class) secondo MedDRA. Le frequenze sono classificate come segue: molto comune (≥1/10), comune (da ≥1/100 a <1/10), non comune (da ≥1/1 000 a <1/100), rara (da ≥1/10 000 a <1/1 000), molto rara (<1/10 000), e non nota (non stimabile dai dati disponibili).
Tabella 5 Frequenza delle reazioni avverse segnalate da studi clinici e sorveglianza post-marketing
|
Classificazione per sistemi e organi (SOC) secondo MedDRA
|
Molto comune
|
Comune
|
Non comune
|
Non nota
|
|
Infezioni e infestazioni
|
|
|
Congiuntivite
Infezione del tratto respiratorio
Cistite
Polmonite
Herpes zoster
Herpes zoster oftalmico
Influenza
Infezioni da stafilococco
Batteriemia streptococcica
|
|
|
Tumori benigni, maligni e non specificati (inclusi cisti e polipi)
|
|
Citopenia refrattaria con displasia multilineare (sindrome mielodisplastica)
|
Leucemia mieloide acuta
Leucemia acuta
Leucemia mielomonocitica cronica
|
|
|
Patologie del sistema emolinfopoietico
|
Trombocitopenia2
Linfopenia3
Anemia4
Pancitopenia
|
Leucopenia5
Neutropenia6
|
Citopenia refrattaria con displasia unilineare
Anemia nefrogenica
Insufficienza midollare
Porpora trombocitopenica
|
|
|
Disturbi del sistema immunitario
|
|
|
Ipersensibilità
|
Angioedema
|
|
Patologie endocrine
|
|
Ipotiroidismo secondario
|
Ipotiroidismo
Diabete mellito
Crisi carcinoide
Iperparatiroidismo
|
|
|
Disturbi del metabolismo e della nutrizione
|
Diminuzione dell'appetito
|
Iperglicemia
Deidratazione
Ipomagnesiemia
Iponatriemia
|
Ipoglicemia
Ipernatremia
Ipofosfatemia
Sindrome da lisi tumorale
Ipercalcemia
Ipocalcemia
Ipoalbuminemia
Acidosi metabolica
|
|
|
Disturbi psichiatrici
|
|
Disturbi del sonno
|
Ansia
Allucinazione
Disorientamento
|
|
|
Patologie del sistema nervoso
|
|
Capogiro
Disgeusia
Mal di testa10
Letargia
Sincope
|
Formicolio
Encefalopatia epatica
Parestesia
Parosmia
Sonnolenza
Compressione del midollo spinale
|
|
|
Patologie dell'occhio
|
|
|
Patologie dell'occhio
|
|
|
Patologie dell'orecchio e del labirinto
|
|
|
Vertigine
|
|
|
Patologie cardiache
|
|
Elettrocardiogramma con prolungamento dell'intervallo QT
|
Fibrillazione atriale
Palpitazioni
Infarto miocardico
Angina pectoris
Shock cardiogeno
|
|
|
Patologie vascolari
|
|
Ipertensione7
Arrossamento cutaneo
Vampate di calore
Ipotensione
|
Vasodilatazione
Estremità fredde
Pallore
Ipotensione ortostatica
Flebite
|
|
|
Patologie respiratorie, toraciche e mediastiniche
|
|
Dispnea
|
Dolore orofaringeo
Versamento pleurico
Aumento dell'espettorato
Sensazione di soffocamento
|
|
|
Patologie gastrointestinali
|
Nausea
Vomito
|
Distensione addominale
Diarrea
Dolore addominale
Costipazione
Dolore addominale superiore
Dispepsia
Gastrite
|
Bocca secca
Flatulenza
Ascite
Dolore gastrointestinale
Stomatite
Ematochezia
Fastidio addominale
Blocco intestinale
Colite
Pancreatite acuta
Emorragia rettale
Melena
Dolore addominale inferiore
Ematemesi
Ascite emorragica
Ileo
|
|
|
Patologie epatobiliari
|
|
Iperbilirubinemia9
|
Diminuzione degli enzimi pancreatici
Lesione epatocellulare
Colestasi
Congestione epatica
Insufficienza epatica
|
|
|
Patologie della cute e del tessuto sottocutaneo
|
|
Alopecia
|
Eruzione cutanea
Secchezza della cute
Gonfiore del viso
Iperidrosi
Prurito diffuso
|
|
|
Patologie del sistema muscoloscheletrico e del tessuto connettivo
|
|
Dolore muscoloscheletrico8
Spasmo muscolare
|
|
|
|
Patologie renali e urinarie
|
|
Danno renale acuto
Ematuria
Insufficienza renale
Proteinuria
|
Leucocituria
Incontinenza urinaria
Diminuzione della velocità di filtrazione glomerulare
Malattia renale
Insufficienza prerenale acuta
Compromissione renale
|
|
|
Patologie generali e condizioni relative alla sede di somministrazione
|
Affaticamento1
|
Reazione al sito di iniezione11
Edema periferico
Dolore al sito di somministrazione
Brividi
Sindrome simil-influenzale
|
Massa al sito di iniezione
Fastidio al torace
Dolore toracico
Piressia
Malessere
Dolore
Decesso
Sensazioni anomale
|
|
|
Esami diagnostici
|
|
Aumento della creatinemia
Aumento di GGT*
Aumento di ALT**
Aumento di AST***
Aumento di ALP**** ematica
|
Diminuzione della potassiemia
Aumento dell'uremia
Aumento dell'emoglobina glicosilata
Diminuzione dell'ematocrito
Proteinuria
Dimagrimento
Aumento della creatinfosfochinasi ematica
Aumento della lattato deidrogenasi ematica
Catecolamine ematiche
Aumento della proteina c-reattiva
|
|
|
Traumatismi, intossicazioni e complicazioni da procedura
|
|
|
Frattura della clavicola
|
|
|
Procedure mediche e chirurgiche
|
|
Trasfusione
|
Drenaggio della cavità addominale
Dialisi
Inserimento di sondino gastrointestinale
Posizionamento di stent
Drenaggio di ascesso
Prelievo di midollo osseo
Polipectomia
|
|
|
Circostanze sociali
|
|
|
Disabilità fisica
|
|
1 Include astenia e affaticamento
2 Include trombocitopenia e diminuzione della conta delle piastrine
3 Include linfopenia e diminuzione della conta dei linfociti
4 Include anemia e diminuzione dell'emoglobina
5 Include leucopenia e diminuzione dei leucociti
6 Include neutropenia e diminuzione della conta dei neutrofili
7 Include ipertensione e crisi ipertensiva
8 Include artralgia, dolore delle estremità, mal di schiena, dolore osseo, dolore al fianco, dolore muscoloscheletrico del torace e dolore al collo
9 Include aumento della bilirubinemia e iperbilirubinemia
10 Include mal di testa ed emicrania
11 Include reazione al sito di iniezione, ipersensibilità al sito di iniezione, indurimento del sito di iniezione, gonfiore del sito di iniezione
*Gamma-glutamiltransferasi
**Alanina aminotransferasi
***Aspartato aminotransferasi
****Fosfatasi alcalina
Descrizione di reazioni avverse selezionate
Mielosoppressione
La tossicità midollare prevalentemente di grado lieve/moderata (mielo/ematossicità) si è manifestata con riduzioni reversibili/transitorie dell'emocromo di tutte le linee emopoietiche (citopenie in tutte le combinazioni, come pancitopenia, bicitopenia, monocitopenia isolata - anemia, neutropenia, linfocitopenia e trombocitopenia). Nonostante sia stata riportata una significativa deplezione selettiva delle cellule B, dopo la radioterapia recettoriale con peptidi marcati (Peptide Receptor Radionuclide Therapy, PRRT) non è stato osservato alcun aumento del tasso di incidenza delle complicanze infettive. A seguito del trattamento con Lutathera, sono stati segnalati casi di patologie ematologiche irreversibili, come neoplasie ematiche premaligne e maligne (ad esempio, rispettivamente, sindrome mielodisplastica e leucemia mieloide acuta).
Nello studio NETTER-1, il nadir piastrinico si è verificato a una mediana di 5,1 mesi dopo la prima dose. Dei 59 pazienti che hanno sviluppato trombocitopenia, il 68% ha avuto un recupero piastrinico ai livelli basali o normali. Il tempo mediano per il recupero piastrinico è stato di 2 mesi. Quindici dei diciannove pazienti in cui il recupero piastrinico non è stato documentato avevano conte piastriniche post-nadir.
Tossicità renale
Lutezio (177Lu) oxodotreotide viene escreto dal rene.
Il progressivo deterioramento a lungo termine della funzione di filtrazione glomerulare dimostrata negli studi clinici, conferma che la nefropatia correlata a Lutathera è una patologia renale cronica che si sviluppa progressivamente per mesi o anni dopo l'esposizione. Prima del trattamento con Lutathera nei pazienti con compromissione renale da lieve e moderata, si raccomanda di valutare il rapporto beneficio-rischio individuale. Per ulteriori dettagli, vedere il paragrafo 4.2 (Tabella 3 e il sottoparagrafo “Compromissione renale”) e il paragrafo 4.4. L'uso di Lutathera è controindicato in pazienti con insufficienza renale con clearance della creatinina <30 mL/min (vedere paragrafo 4.3).
Crisi ormonali neuroendocrine
Raramente sono state osservate crisi ormonali correlate al rilascio di sostanze bioattive (probabilmente dovute a lisi di cellule tumorali neuroendocrine), che comunque si sono risolte dopo adeguato trattamento medico (vedere paragrafo 4.4).
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l'autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite il sistema nazionale di segnalazione dell'Agenzia Italiana del Farmaco, Sito web: www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Sovradosaggio
Cosa fare se avete preso una dose eccessiva di Lutathera
Il sovradosaggio con Lutathera è improbabile poiché questo medicinale viene fornito come prodotto a singola dose e pronto all'uso contenente una quantità di radioattività predefinita ed è somministrato da personale autorizzato a maneggiare radiofarmaci dopo la valutazione del paziente da parte di un medico qualificato. In caso di sovradosaggio è prevedibile un aumento della frequenza delle reazioni avverse correlate a radiotossicità.
In caso di somministrazione di un sovradosaggio di radiazioni con Lutathera, ove possibile, la dose assorbita dal paziente deve essere ridotta stimolando l'escrezione corporea del radionuclide tramite diuresi forzata e frequente svuotamento della vescica nel corso delle prime 48 ore dopo l'infusione. In tali circostanze, può essere utile calcolare l'effettiva dose applicata.
Ogni settimana, nelle successive 10 settimane, devono essere effettuati i seguenti esami di laboratorio:
- Monitoraggio ematologico: conta dei globuli bianchi con conte differenziali, piastrine ed emoglobina
- Monitoraggio ematochimico: creatinina sierica e glicemia.
Scadenza
72 ore dalla data e ora di calibrazione.
Conservazione
Conservare a temperatura inferiore a 25°C.
Non congelare.
Conservare nella confezione originale per la protezione anti-radiazioni ionizzanti (schermatura di piombo).
La conservazione dei radiofarmaci deve essere conforme alle normative nazionali sui materiali radioattivi.
Foglietto Illustrativo
Fonti Ufficiali
Servizi Avanzati



© 2022 EDRA S.p.A. - P.iva 08056040960
DPO - dpo@lswr.it