
Jemperli
Ultimo aggiornamento: 19/04/2024
Cos'è Jemperli?
Jemperli è un farmaco a base del principio attivo
Dostarlimab, appartenente alla categoria degli
Antineoplastici e nello specifico
Inibitori di PD-1/PDL-1 (prot. morte cellulare prog. 1/ligand 1). E' commercializzato in Italia dall'azienda
GlaxoSmithKline S.p.A..
Jemperli può essere prescritto con Ricetta OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile.
Jemperli può essere prescritto con Ricetta OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile.
Confezioni
Jemperli 500 mg soluzione per infusione uso endovenoso 1 flaconcino 10 ml (50 mg/ml)
Informazioni commerciali sulla prescrizione
Titolare: GlaxoSmithKline (Ireland) Limited
Concessionario: GlaxoSmithKline S.p.A.
Ricetta: OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile
Classe: H
Principio attivo: Dostarlimab
Gruppo terapeutico: Antineoplastici
ATC: L01FF07 - Dostarlimab
Forma farmaceutica: soluzione (uso interno)
Concessionario: GlaxoSmithKline S.p.A.
Ricetta: OSP - medicinali soggetti a prescrizione medica limitativa, utilizzabili esclusivamente in ambiente ospedaliero o in struttura ad esso assimilabile
Classe: H
Principio attivo: Dostarlimab
Gruppo terapeutico: Antineoplastici
ATC: L01FF07 - Dostarlimab
Forma farmaceutica: soluzione (uso interno)
Se sei un professionista, potrai trovare le schede tecniche complete e molto altro nell'area riservata di Codifa.it
Indicazioni
Perché si usa Jemperli? A cosa serve?
JEMPERLI è indicato in associazione a carboplatino e paclitaxel per il trattamento di pazienti adulte affette da cancro endometriale (CE) primario avanzato o ricorrente con deficit del sistema di mismatch repair (dMMR)/elevata instabilità dei microsatelliti (MSI-H) e che sono candidate per la terapia sistemica.
JEMPERLI è indicato come monoterapia per il trattamento di pazienti adulte affette da CE avanzato o ricorrente dMMR/MSI-H, progredito durante o dopo un precedente trattamento con un regime a base di platino.
Posologia
Come usare Jemperli: Posologia
La terapia deve essere iniziata e monitorata da medici specialisti, esperti nel trattamento del cancro.
La determinazione dello stato dMMR/MSI-H del tumore deve essere effettuata utilizzando un test convalidato, quale IHC, PCR oppure NGS* (vedere paragrafo 5.1 per informazioni sui test utilizzati negli studi).
*IHC = immunoistochimica; PCR = reazione a catena della polimerasi; NGS = sequenziamento di nuova generazione.
Posologia
JEMPERLI in associazione a carboplatino e paclitaxel
Quando JEMPERLI è somministrato in associazione a carboplatino e paclitaxel, fare riferimento ai Riassunti delle Caratteristiche del Prodotto completi dei medicinali usati in combinazione (vedere anche paragrafo 5.1).
La dose raccomandata è di 500 mg di Dostarlimab in associazione a carboplatino e paclitaxel ogni 3 settimane per 6 cicli, seguita da 1 000 mg di dostarlimab in monoterapia ogni 6 settimane per tutti i cicli successivi.
Il regime posologico in associazione a carboplatino e paclitaxel è illustrato nella Tabella 1.
Tabella 1. Regime posologico per JEMPERLI in associazione a carboplatino e paclitaxel
Tabella 1. Regime posologico per JEMPERLI in associazione a carboplatino e paclitaxel
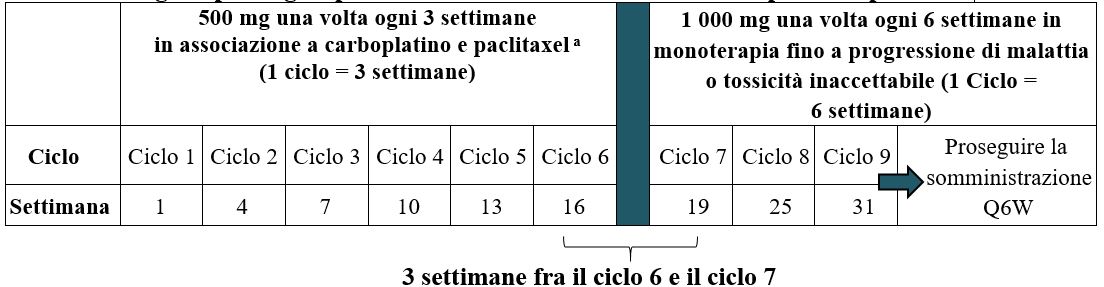
a Somministrare dostarlimab prima di carboplatino e paclitaxel nello stesso giorno.
La somministrazione di dostarlimab deve proseguire secondo lo schema raccomandato fino a progressione di malattia o tossicità inaccettabile o per una durata fino a 3 anni (vedere paragrafo 5.1).
JEMPERLI in monoterapia
La dose raccomandata in monoterapia è di 500 mg di dostarlimab ogni 3 settimane per 4 cicli, seguita da 1 000 mg ogni 6 settimane per tutti i cicli successivi.
Il regime posologico in monoterapia è illustrato nella Tabella 2.
Tabella 2. Regime posologico per JEMPERLI in monoterapia
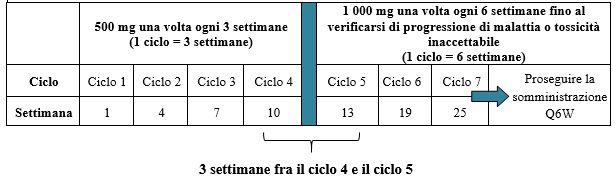
La somministrazione di dostarlimab deve proseguire secondo lo schema raccomandato fino a progressione di malattia o tossicità inaccettabile (vedere paragrafo 5.1).
Modifiche della dose
Non è raccomandata una riduzione della dose. Può essere necessario posticipare la somministrazione o interrompere il trattamento in base alla sicurezza e alla tollerabilità individuali. Le modifiche raccomandate per la gestione delle reazioni avverse sono indicate nella Tabella 3.
Le linee guida dettagliate per il trattamento delle reazioni avverse immuno-correlate e delle reazioni correlate a infusione sono descritte nel paragrafo 4.4.
Tabella 3. Modifiche raccomandate della dose di JEMPERLI
|
Reazioni avverse immuno-correlate
|
Grado di severitàa
|
Modifica della dose
|
|
Colite
|
2 o 3
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1.
|
|
4
|
Interrompere la somministrazione in modo permanente.
|
|
|
Epatite
|
Grado 2 con ASTb o ALTc > 3 e fino a 5 × ULNd
oppure bilirubina totale > 1,5 e fino a 3 × ULN
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1.
|
|
Grado ≥ 3 con AST o ALT > 5 × ULN
oppure bilirubina totale > 3 × ULN
|
Interrompere la somministrazione in modo permanente (vedere l'eccezione indicata di seguito)e.
|
|
|
Diabete mellito di tipo 1 (DMT1)
|
3 o 4 (iperglicemia)
|
Sospendere la somministrazione. Riprendere la somministrazione nelle pazienti stabili dal punto di vista clinico e metabolico, opportunamente gestite.
|
|
Ipofisite o insufficienza surrenalica
|
2, 3 o 4
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1. Interrompere la somministrazione in modo permanente in caso di recidiva o peggioramento in corso di adeguata terapia ormonale.
|
|
Ipotiroidismo o ipertiroidismo
|
3 o 4
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1.
|
|
Polmonite
|
2
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1. In caso di recidiva di grado 2, interrompere la somministrazione in modo permanente.
|
|
3 o 4
|
Interrompere la somministrazione in modo permanente.
|
|
|
Nefrite
|
2
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1.
|
|
3 o 4
|
Interrompere la somministrazione in modo permanente.
|
|
|
Condizioni dermatologiche esfoliative (es. SJS, TEN, DRESS)
|
Sospetto
|
Sospendere la somministrazione qualsiasi sia il grado. Riprendere la somministrazione se non confermato e in seguito a risoluzione della tossicità a grado 0 o 1.
|
|
Confermato
|
Interrompere la somministrazione in modo permanente.
|
|
|
Miocardite
|
2, 3 o 4
|
Interrompere la somministrazione in modo permanente.
|
|
Tossicità neurologiche severe (sindrome miastenica/miastenia gravis, sindrome di Guillain-Barré, encefalite, mielite trasversa)
|
2, 3 o 4
|
Interrompere la somministrazione in modo permanente.
|
|
Altre reazioni avverse immuno-correlate (incluse ma non limitate a miosite, sarcoidosi, anemia emolitica autoimmune, pancreatite, iridociclite, uveite, chetoacidosi diabetica, artralgia, rigetto di trapianto di organo solido, malattia da trapianto contro l'ospite)
|
3
|
Sospendere la somministrazione. Riprendere la somministrazione in seguito a risoluzione della tossicità a grado 0 o 1.
|
|
4
|
Interrompere la somministrazione in modo permanente.
|
|
|
Recidiva di reazioni avverse immuno-correlate in seguito a risoluzione a ≤ grado 1 (ad eccezione della polmonite, vedere sopra)
|
3 o 4
|
Interrompere la somministrazione in modo permanente.
|
|
Reazioni da infusione
|
2
|
Sospendere la somministrazione. In caso di risoluzione entro un'ora dall'interruzione, è possibile riprendere la somministrazione al 50% della velocità di infusione originale, oppure riprendere in seguito a risoluzione dei sintomi con la premedicazione. In caso di recidiva di grado 2 nonostante un'adeguata premedicazione, interrompere la somministrazione in modo permanente.
|
|
3 o 4
|
Interrompere la somministrazione in modo permanente.
|
a Tossicità classificata secondo i criteri terminologici comuni per gli eventi avversi (CTCAE, Common Terminology Criteria for Adverse Events) del National Cancer Institute, versione 5.0.
b AST = aspartato aminotransferasi
c ALT = alanina aminotransferasi
d ULN = limite superiore della norma
e Per le pazienti con metastasi epatiche che iniziano il trattamento con un aumento di AST o ALT di grado 2, il trattamento dovrà essere interrotto in caso di incremento di AST o ALT ≥ 50% rispetto al basale protratto per almeno 1 settimana.
Scheda paziente
Tutti i medici che prescrivono JEMPERLI devono informare le pazienti in merito alla Scheda paziente, spiegando loro come comportarsi qualora dovessero manifestare qualsiasi sintomo di reazioni avverse immuno-correlate. Il medico consegnerà una Scheda paziente a ciascuna paziente.
Popolazioni speciali
Anziani
Non sono raccomandati aggiustamenti della dose nei pazienti di età pari o superiore a 65 anni.
Vi sono dati clinici limitati relativi all'uso di dostarlimab in pazienti di età pari o superiore a 75 anni (vedere paragrafo 5.1).
Compromissione renale
Non sono raccomandati aggiustamenti della dose nelle pazienti con compromissione renale lieve o moderata. Vi sono dati limitati su pazienti con compromissione renale severa o patologia renale allo stadio terminale sottoposte a dialisi (vedere paragrafo 5.2).
Compromissione epatica
Non sono raccomandati aggiustamenti della dose nelle pazienti con compromissione epatica lieve. Vi sono dati limitati su pazienti con compromissione epatica moderata e non sono disponibili dati su pazienti con compromissione epatica severa (vedere paragrafo 5.2).
Popolazione pediatrica
La sicurezza e l'efficacia di JEMPERLI nei bambini e negli adolescenti di età inferiore a 18 anni non sono state stabilite. Non ci sono dati disponibili.
Modo di somministrazione
JEMPERLI è destinato esclusivamente all'infusione endovenosa. JEMPERLI deve essere somministrato tramite infusione endovenosa della durata di 30 minuti utilizzando una pompa per infusione endovenosa.
JEMPERLI non deve essere somministrato per via endovenosa rapida o con somministrazione in bolo.
Per le istruzioni sulla diluizione del medicinale prima della somministrazione, vedere paragrafo 6.6.
Controindicazioni
Quando non dev'essere usato Jemperli
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
Avvertenze speciali e precauzioni di impiego
Cosa serve sapere prima di prendere Jemperli
Tracciabilità
Al fine di migliorare la tracciabilità dei medicinali biologici, il nome commerciale e il numero di lotto del medicinale somministrato devono essere chiaramente registrati.
Reazioni avverse immuno-correlate
Nei pazienti trattati con anticorpi che bloccano la via della proteina di morte cellulare programmata-1 / del ligando di morte cellulare programmata-1 (PD-1/PD-L1), incluso Dostarlimab, è possibile che si verifichino reazioni avverse immuno-correlate, che possono essere severe o fatali. Benché, generalmente, le reazioni avverse immuno-correlate si presentino durante il trattamento con anticorpi anti-PD-1/PD-L1, i sintomi possono manifestarsi anche dopo l'interruzione del trattamento. Le reazioni avverse immuno-correlate possono interessare qualsiasi organo o tessuto e coinvolgere più di un apparato/sistema contemporaneamente. Le reazioni avverse immuno-correlate importanti elencate in questo paragrafo non comprendono tutte le possibili reazioni avverse immuno-correlate severe o fatali.
L'identificazione e la gestione tempestive delle reazioni avverse immuno-correlate sono fondamentali per garantire un utilizzo sicuro degli anticorpi anti-PD-1/PD-L1. I pazienti devono essere monitorati per l'eventuale presenza di segni e sintomi di reazioni avverse immuno-correlate. I parametri ematologici e chimico-clinici, inclusi i test di funzionalità epatica, renale e tiroidea, devono essere valutati al basale e periodicamente durante il trattamento. Per le sospette reazioni avverse immuno-correlate, si deve garantire un'adeguata valutazione, compreso un consulto specialistico.
In base alla severità della reazione avversa, il trattamento con dostarlimab deve essere sospeso o interrotto in modo permanente e si devono somministrare corticosteroidi (da 1 a 2 mg/kg/die di prednisone o equivalente) o altra terapia appropriata (vedere di seguito e paragrafo 4.2). Al miglioramento dei sintomi a Grado ≤ 1, iniziare una riduzione graduale dei corticosteroidi, continuandola per almeno 1 mese. In base a dati limitati emersi dagli studi clinici su pazienti nei quali non è stato possibile ottenere il controllo delle reazioni avverse immuno-correlate con l'uso di corticosteroidi, può essere valutata la somministrazione di altri immunosoppressori sistemici. Se necessario, istituire una terapia sostitutiva ormonale per le endocrinopatie.
Il trattamento con dostarlimab deve essere interrotto definitivamente in presenza di una qualsiasi reazione avversa immuno-correlata di Grado 3 recidivante e in presenza di una qualsiasi tossicità per reazioni avverse immuno-correlate di Grado 4, ad eccezione delle endocrinopatie controllate tramite terapia ormonale sostitutiva e salvo diversamente specificato nella Tabella 3.
Polmonite immuno-correlata
Nei pazienti in trattamento con dostarlimab è stata segnalata polmonite (vedere paragrafo 4.8). I pazienti devono essere monitorati per rilevare l'insorgenza di segni e sintomi di polmonite. I casi di sospetta polmonite devono essere confermati tramite indagini radiologiche e devono essere escluse altre cause. I pazienti devono essere gestiti con modifiche del trattamento con dostarlimab e con somministrazione di corticosteroidi (vedere paragrafo 4.2).
Colite immuno-correlata
Dostarlimab può causare colite immuno-correlata (vedere paragrafo 4.8). I pazienti devono essere monitorati per rilevare l'insorgenza di segni e sintomi di colite e devono essere gestiti con modifiche del trattamento con dostarlimab, con somministrazione di antidiarroici e con corticosteroidi (vedere paragrafo 4.2).
Epatite immuno-correlata
Dostarlimab può causare epatite immuno-correlata (vedere paragrafo 4.8). I pazienti devono essere monitorati periodicamente per rilevare alterazioni della funzionalità epatica secondo quanto indicato, sulla base della valutazione clinica, e devono essere gestiti con modifiche del trattamento con dostarlimab e con somministrazione di corticosteroidi (vedere paragrafo 4.2).
Endocrinopatie immuno-correlate
Nei pazienti in trattamento con dostarlimab sono state segnalate endocrinopatie immuno-correlate, tra cui ipotiroidismo, ipertiroidismo, tiroidite, ipofisite, diabete mellito di tipo 1, chetoacidosi diabetica e insufficienza surrenalica (vedere paragrafo 4.8).
Ipotiroidismo e ipertiroidismo
Nei pazienti in trattamento con dostarlimab si sono verificati ipotiroidismo e ipertiroidismo immuno-correlati (compresa tiroidite) e l'ipotiroidismo può presentarsi successivamente all'ipertiroidismo. I pazienti devono essere monitorati per rilevare risultati anomali dei test della funzionalità tiroidea prima di iniziare la terapia e periodicamente durante il trattamento, e come indicato in base alla valutazione clinica. L'ipotiroidismo e l'ipertiroidismo immuno-correlati (compresa tiroidite) devono essere gestiti come raccomandato nel paragrafo 4.2.
Insufficienza surrenalica
Nei pazienti in trattamento con dostarlimab si è verificata insufficienza surrenalica immuno-correlata. I pazienti devono essere monitorati per rilevare l'insorgenza di segni clinici e sintomi di insufficienza surrenalica. In caso di insufficienza surrenalica sintomatica, i pazienti devono essere gestiti come raccomandato nel paragrafo 4.2.
Nefrite immuno-correlata
Dostarlimab può causare nefrite immuno-correlata (vedere paragrafo 4.8). I pazienti devono essere monitorati per rilevare alterazioni della funzionalità renale e gestiti con modifiche del trattamento con dostarlimab e con somministrazione di corticosteroidi (vedere paragrafo 4.2).
Eruzione cutanea immuno-correlata
Nei pazienti in trattamento con dostarlimab sono state segnalate eruzioni cutanee immuno-correlate, compreso pemfigoide (vedere paragrafo 4.8). I pazienti devono essere monitorati per rilevare l'insorgenza di segni e sintomi di eruzione cutanea. Le condizioni dermatologiche esfoliative devono essere gestite come raccomandato nel paragrafo 4.2. In pazienti trattati con inibitori di PD-1 sono stati segnalati casi di Sindrome di Stevens-Johnson o necrolisi tossica epidermica.
Deve essere usata cautela nel valutare l'utilizzo di dostarlimab in un paziente che abbia in precedenza manifestato una reazione avversa cutanea severa o potenzialmente fatale durante un precedente trattamento con altri agenti antitumorali immunostimolanti.
Artralgia immuno-correlata
Nei pazienti in trattamento con dostarlimab è stata segnalata artralgia immuno-correlata (vedere paragrafo 4.8). I pazienti devono essere monitorati per rilevare segni e sintomi di artralgia. Una sospetta artralgia immuno-correlata deve essere confermata e devono essere escluse altre cause. I pazienti devono essere gestiti con modifiche del trattamento con dostarlimab e con somministrazione di corticosteroidi (vedere paragrafo 4.2).
Altre reazioni avverse immuno-correlate
Considerato il meccanismo d'azione di dostarlimab, possono verificarsi altre potenziali reazioni avverse immuno-correlate, inclusi eventi potenzialmente gravi [ad es. miosite, miocardite, encefalite, neuropatia demielinizzante (inclusa sindrome di Guillain-Barré), sarcoidosi]. Le reazioni avverse immuno-correlate clinicamente significative segnalate in meno dell'1% dei pazienti in trattamento con dostarlimab in monoterapia negli studi clinici comprendono encefalite, anemia emolitica autoimmune, pancreatite, iridociclite e uveite. I pazienti devono essere monitorati per rilevare l'insorgenza di segni e sintomi di reazioni avverse immuno-correlate e devono essere gestiti come descritto nel paragrafo 4.2. In pazienti in trattamento con inibitori di PD-1 sono stati segnalati casi di rigetto di trapianto di organo solido nella fase successiva all'immissione in commercio. Il trattamento con dostarlimab può aumentare il rischio di rigetto nei soggetti sottoposti a trapianto di organo solido. In questi pazienti deve essere valutato il beneficio del trattamento con dostarlimab rispetto al rischio di possibile rigetto di organo.
Altre complicanze gravi o fatali possono insorgere in pazienti sottoposti a trapianto allogenico di cellule staminali ematopoietiche (HSCT) prima o dopo il trattamento con un anticorpo anti-PD-1/PD-L1. Le complicanze correlate al trapianto comprendono malattia del trapianto contro l'ospite (GvHD) iperacuta, GvHD acuta, GvHD cronica, patologia veno-occlusiva epatica in seguito a condizionamento a intensità ridotta, e sindrome febbrile con necessità di trattamento steroideo (senza una causa infettiva identificata). Tali complicanze possono insorgere nonostante terapia intermedia tra il blocco PD-1/PD-L1 e l'HSCT allogenico. I pazienti dovranno essere monitorati attentamente per rilevare evidenze di complicanze correlate al trapianto e istituire un intervento tempestivo. Valutare il beneficio rispetto ai rischi del trattamento con un anticorpo anti-PD-1/PD-L1 prima o dopo HSCT allogenico.
Reazioni correlate a infusione
Dostarlimab può causare reazioni da infusione, che possono essere severe (vedere paragrafo 4.8). In caso di reazioni da infusione severe (di grado 3) o potenzialmente fatali (di grado 4), sospendere l'infusione e interrompere definitivamente il trattamento (vedere paragrafo 4.2).
Pazienti esclusi dagli studi clinici
Sono stati esclusi dallo studio GARNET i pazienti che presentavano le seguenti condizioni: performance score (PS) secondo Eastern Cooperative Oncology Group (ECOG) al basale ≥ 2; metastasi non controllate del sistema nervoso centrale o meningite carcinomatosa; altre neoplasie maligne nel corso degli ultimi 2 anni; immunodeficienza o somministrazione di terapia immunosoppressiva nei 7 giorni precedenti; infezione attiva da HIV, epatite B o epatite C; patologia autoimmune attiva che abbia richiesto un trattamento sistemico negli ultimi 2 anni, a eccezione della terapia sostitutiva; anamnesi di pneumopatia interstiziale; o somministrazione di un vaccino vivo nei 14 giorni precedenti.
Contenuto di sodio
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per dose da 500 mg, cioè essenzialmente “senza sodio”.
Interazioni con altri medicinali e altre forme di interazione
Quali farmaci o alimenti possono modificare l'effetto di Jemperli
Non sono stati effettuati studi d'interazione. Gli anticorpi monoclonali (mAb) quali dostarlimab non sono substrati del citocromo P450 o dei trasportatori dei principi attivi. Dostarlimab non è una citochina ed è improbabile che agisca come modulatore delle citochine. Inoltre, non sono attese interazioni farmacocinetiche (PK) tra dostarlimab e principi attivi di piccole dimensioni molecolari. Non vi sono evidenze di interazioni mediate da clearance non specifica della degradazione lisosomiale per gli anticorpi.
Fertilità, gravidanza e allattamento
Donne in età fertile/Contraccezione
Vi è un rischio associato alla somministrazione di Dostarlimab per le donne potenzialmente fertili. Le donne in età fertile devono usare misure contraccettive efficaci durante il trattamento con dostarlimab e fino a 4 mesi dopo l'ultima dose di dostarlimab.
Gravidanza
I dati relativi all'uso di dostarlimab in donne in gravidanza non esistono o sono in numero limitato. Sulla base del meccanismo d'azione, dostarlimab può causare effetti farmacologici dannosi sul feto quando somministrato durante la gravidanza.
Non sono stati condotti studi con dostarlimab sullo sviluppo o sulla riproduzione negli animali; tuttavia, l'inibizione della via PD-1/PD-L1 può aumentare il rischio di rigetto immuno-correlato del feto in via di sviluppo, con conseguente morte fetale (vedere paragrafo 5.3). Come noto, le immunoglobuline umane (IgG4) attraversano la barriera placentare. Pertanto, è possibile che dostarlimab, essendo una IgG4, venga trasmesso dalla madre al feto in via di sviluppo.
JEMPERLI non è raccomandato durante la gravidanza e in donne in età fertile che non usano misure contraccettive efficaci.
Allattamento
Non è noto se dostarlimab/metaboliti siano escreti nel latte materno.
Il rischio per i neonati/lattanti non può essere escluso.
JEMPERLI non deve essere utilizzato durante l'allattamento e l'allattamento deve essere evitato per almeno 4 mesi dopo l'ultima dose di dostarlimab.
Fertilità
Non sono stati condotti studi di fertilità con dostarlimab (vedere paragrafo 5.3).
Effetti sulla capacità di guidare veicoli e sull'uso di macchinari
JEMPERLI non altera o altera in modo trascurabile la capacità di guidare veicoli e di usare macchinari.
Effetti indesiderati
Quali sono gli effetti collaterali di Jemperli
Riassunto del profilo di sicurezza
Dostarlimab è generalmente associato a reazioni avverse immuno-correlate. La maggior parte di queste, comprese le reazioni severe, si è risolta con l'impiego di una terapia medica appropriata o la sospensione di dostarlimab (vedere di seguito “Descrizione di reazioni avverse selezionate”).
Dostarlimab in monoterapia
La sicurezza di dostarlimab è stata valutata in 605 pazienti con CE o altri tumori solidi in stadio avanzato trattati con dostarlimab in monoterapia nel corso dello studio GARNET, fra i quali 153 pazienti affette da CE dMMR/MSI-H avanzato o recidivante. I pazienti hanno ricevuto dosi da 500 mg ogni 3 settimane per 4 cicli, seguite da 1 000 mg ogni 6 settimane per tutti i cicli successivi.
Nei pazienti con tumori solidi avanzati o recidivanti (N = 605), le reazioni avverse più comuni (> 10%) sono state anemia (28,6%), diarrea (26,0%), nausea (25,8%), vomito (19,0%), artralgia (17,0%), prurito (14,2%), eruzione cutanea (13,2%), piressia (12,4%), aspartato aminotransferasi aumentata (11,2%) e ipotiroidismo (11,2%). Il trattamento con JEMPERLI è stato interrotto definitivamente a causa delle reazioni avverse in 38 pazienti (6,3%). Per la maggior parte, si è trattato di eventi immuno-correlati. Si sono verificate reazioni avverse gravi nell'11,2% delle pazienti; le reazioni avverse più gravi sono state reazioni avverse immuno-correlate (vedere paragrafo 4.4).
Il profilo di sicurezza per le pazienti con CE dMMR/MSI-H nello studio GARNET (N = 153) non è stato diverso da quello della popolazione complessiva in monoterapia presentato in Tabella 4.
Dostarlimab in associazione a carboplatino e paclitaxel
La sicurezza di dostarlimab è stata valutata in 241 pazienti con CE primario avanzato o ricorrente che hanno ricevuto dostarlimab in associazione a carboplatino e paclitaxel nello studio RUBY. Le pazienti hanno ricevuto dosi da 500 mg di dostarlimab ogni 3 settimane per 6 cicli, seguite da 1 000 mg ogni 6 settimane per tutti i cicli successivi.
Nelle pazienti con CE primario avanzato o ricorrente (N = 241), le reazioni avverse più comuni (> 10%) sono state eruzione cutanea (22,8%), eruzione cutanea maculopapulare (14,1%), ipotiroidismo (14,1%), alanina aminotransferasi aumentate (12,9%), aspartato aminotransferasi aumentate (12,0%), piressia (12,0%) e cute secca (10,4%). Il trattamento con JEMPERLI è stato interrotto definitivamente a causa delle reazioni avverse in 12 (5%) pazienti, la maggior parte erano eventi immuno-correlati. Si sono verificate reazioni avverse gravi nel 5,8% delle pazienti; le reazioni avverse più gravi sono state reazioni avverse immuno-correlate (vedere paragrafo 4.4).
Nello studio RUBY il profilo di sicurezza per le pazienti con CE dMMR/MSI-H (N=52) non è stato diverso da quello della popolazione complessiva (N=241) presentato nella Tabella 4.
Tabella delle reazioni avverse
Le reazioni avverse segnalate nelle sperimentazioni cliniche di dostarlimab in monoterapia o in associazione a chemioterapia sono elencate nella Tabella 4 secondo la classificazione per sistemi e organi e per frequenza. Le frequenze delle reazioni avverse elencate nella colonna relativa a dostarlimab in monoterapia sono basate sulla frequenza degli eventi avversi da tutte le cause, identificati in 605 pazienti affetti da tumori solidi avanzati o recidivanti dello studio GARNET esposti a monoterapia con dostarlimab per una durata mediana di trattamento di 24 settimane (intervallo (range): da 1 settimana a 229 settimane). Se non diversamente indicato, le frequenze delle reazioni avverse elencate nella colonna relativa a dostarlimab in associazione a chemioterapia sono basate sulla frequenza degli eventi avversi da tutte le cause, identificati in 241 pazienti con CE primario avanzato o ricorrente dello studio RUBY esposte a dostarlimab in associazione a carboplatino e paclitaxel per una durata mediana di trattamento di 43 settimane (range: da 3 a 151 settimane). Per ulteriori informazioni di sicurezza nel caso di somministrazione di dostarlimab in associazione a carboplatino e paclitaxel, per i medicinali concomitanti fare riferimento al rispettivo Riassunto delle Caratteristiche del Prodotto.
Le reazioni avverse che è noto potrebbero manifestarsi con dostarlimab in monoterapia o con carboplatino o paclitaxel somministrati singolarmente, potrebbero verificarsi durante il trattamento di combinazione dei tre farmaci, anche nel caso che queste reazioni avverse non risultino precedentemente segnalate negli studi clinici con dostarlimab in associazione a carboplatino e paclitaxel.
Queste reazioni sono elencate secondo la classificazione per sistemi e organi e in base alla frequenza. Le categorie di frequenza sono le seguenti: molto comune (≥ 1/10), comune (≥ 1/100, < 1/10), non comune (≥ 1/1 000, < 1/100), raro (≥ 1/10 000, < 1/1 000), molto raro (< 1/10 000) e non nota (la frequenza non può essere definita sulla base dei dati disponibili).
Tabella 4: Reazioni avverse nelle pazienti trattate con dostarlimab
|
|
Dostarlimab in monotherapia
|
Dostarlimab in associazione a chemioterapia
|
|
Patologie del sistema emolinfopoietico
|
||
|
Molto comune
|
Anemiaa
|
|
|
Patologie endocrine
|
||
|
Molto comune
|
Ipotiroidismo*b
|
Ipotiroidismo e
|
|
Comune
|
Ipertiroidismo*, insufficienza surrenalica *
|
Ipertiroidismo, insufficienza surrenalica
|
|
Non comune
|
Tiroidite*c, ipofisited
|
Tiroidite
|
|
Disturbi del metabolismo e della nutrizione
|
||
|
Non comune
|
Diabete mellito di tipo 1, chetoacidosi diabetica
|
Diabete mellito di tipo 1
|
|
Patologie del sistema nervoso
|
||
|
Non comune
|
Encefalite, miastenia gravis
|
Sindrome miastenica f
|
|
Patologie dell'occhio
|
||
|
Non comune
|
Uveiteg
|
Uveite
|
|
Patologie cardiache
|
||
|
Non comune
|
|
Miocarditeh
|
|
Patologie respiratorie, toraciche e mediastiniche
|
||
|
Comune
|
Polmonite*i
|
Polmonite
|
|
Patologie gastrointestinali
|
||
|
Molto comune
|
Diarrea, nausea,
vomito
|
|
|
Comune
|
Colite *j, pancreatitek, gastrite
|
Colitel
|
|
Non comune
|
Esofagite
|
Pancreatite, gastrite immuno-mediataf, vasculite gastrointestinalef
|
|
Patologie epatobiliari
|
||
|
Comune
|
Epatite*m
|
|
|
Patologie della cute e del tessuto sottocutaneo
|
||
|
Molto comune
|
Eruzione cutanea*n, prurito
|
Eruzione cutaneao, cute secca
|
|
Patologie del sistema muscoloscheletrico e del tessuto connettivo
|
||
|
Molto comune
|
Artralgia*
|
|
|
Comune
|
Mialgia
|
|
|
Non comune
|
Artrite immuno-mediata, polimialgia reumatica, miosite immuno-mediata
|
Artrite immuno-mediata, miositep
|
|
Patologie renali e urinarie
|
||
|
Non comune
|
Nefrite*q
|
|
|
Patologie generali e condizioni relative alla sede di somministrazione
|
||
|
Molto comune
|
Piressia
|
Piressia
|
|
Comune
|
Brividi
|
|
|
Non comune
|
|
Sindrome da risposta infiammatoria sistemicap
|
|
Esami diagnostici
|
||
|
Molto comune
|
Transaminasi aumentater
|
Alanina aminotransferasi aumentate, aspartato aminotransferasi aumentate
|
|
Traumatismi, intossicazioni e complicazioni da procedura
|
||
|
Comune
|
Reazione da infusione*s
|
|
* Vedere paragrafo “Descrizione di reazioni avverse selezionate”
a Include anemia e anemia emolitica autoimmune
b Include ipotiroidismo e ipotiroidismo autoimmune
c Include tiroidite e tiroidite autoimmune
d Include ipofisite e ipofisite linfocitica
e Include ipotiroidismo e ipotiroidismo immuno-mediato
f Segnalato da studi clinici in corso in cieco di dostarlimab in combinazione; categoria di frequenza stimata
gInclude uveite e iridociclite
h Include miocardite (combinazione con chemioterapia) e miocardite immuno-mediata da studi clinici in corso in cieco di dostarlimab in combinazione; categoria di frequenza stimata
i Include polmonite, pneumopatia interstiziale e malattia polmonare immuno-mediata
j Include colite, enterocolite ed enterocolite immuno-mediata
k Include pancreatite e pancreatite acuta
l Include colite (combinazione con chemioterapia) ed enterite segnalate da studi clinici in corso di dostarlimab in combinazione
m Include epatite, epatite autoimmune e citolisi epatica
n Include eruzione cutanea, eruzione cutanea maculo-papulare, eritema, eruzione cutanea maculare, eruzione cutanea pruriginosa, eruzione eritematosa, eruzione cutanea papulare, eritema multiforme, tossicità cutanea, eruzione da farmaco, eruzione cutanea tossica, eruzione esfoliativa e pemfigoide
o Include eruzione cutanea ed eruzione cutanea maculo-papulare
p Segnalata in uno studio clinico in corso di dostarlimab in combinazione
q Include nefrite e nefrite tubulo-interstiziale
r Include transaminasi aumentate, alanina aminotransferasi aumentate, aspartato aminotransferasi aumentate e ipertransaminasemia
s Include reazioni da infusione e ipersensibilità
Descrizione di reazioni avverse selezionate
Le reazioni avverse selezionate descritte di seguito si basano sui dati di sicurezza di dostarlimab in monoterapia in pazienti affetti da CE o altri tumori solidi in fase avanzata, secondo un database combinato su una popolazione di 605 pazienti arruolati nello studio GARNET. Le reazioni avverse immuno-correlate erano definite come eventi di grado 2 e superiore; le frequenze riportate di seguito escludono gli eventi di grado 1. Le linee guida per la gestione di queste reazioni avverse sono illustrate nel paragrafo 4.2.
Reazioni avverse immuno-correlate (vedere paragrafo 4.4)
Polmonite immuno-correlata
Si è verificata polmonite immuno-correlata in 14 pazienti (2,3%), compresi casi di polmonite di grado 2 (1,3%), di grado 3 (0,8%) e di grado 4 (0,2%). La polmonite ha portato all'interruzione di dostarlimab in 8 pazienti (1,3%).
L'uso di corticosteroidi sistemici (≥ 40 mg di prednisone/die o equivalente) si è reso necessario in 11 pazienti (78,6%) che avevano manifestato polmonite. La polmonite si è risolta in 11 pazienti (78,6%).
Colite immuno-correlata
Si è verificata colite in 8 pazienti (1,3%), compresi casi di colite di grado 2 (0,7%) e di grado 3 (0,7%). La colite non ha comportato l'interruzione di dostarlimab in alcuno dei pazienti.
L'uso di corticosteroidi sistemici (≥ 40 mg di prednisone/die o equivalente) si è reso necessario in 5 pazienti (62,5%). La colite si è risolta in 5 (62,5%) dei pazienti che avevano manifestato colite.
Epatite immuno-correlata
Si è verificata epatite in 3 pazienti (0,5%), in tutti i casi di grado 3. Sono stati necessari corticosteroidi sistemici (prednisone ≥ 40 mg/die o equivalente) in 2 pazienti (66,7%). L'epatite ha portato a interruzione di dostarlimab in 1 paziente (0,2%) e si è risolta in 2 su 3 pazienti.
Endocrinopatie immuno-correlate
Si è verificato ipotiroidismo in 46 pazienti (7,6%), tutti di grado 2. L'ipotiroidismo non ha portato a interruzione di dostarlimab e si è risolto in 17 pazienti (37,0%).
Si è verificato ipertiroidismo in 14 pazienti (2,3%), che ha incluso casi di grado 2 (2,1%) e di grado 3 (0,2%). L'ipertiroidismo non ha portato a interruzione di dostarlimab e si è risolto in 10 pazienti (71,4%).
Si è verificata tiroidite in 3 pazienti (0,5%), in tutti i casi di grado 2. Nessuno dei casi di tiroidite si è risolto; non vi sono state interruzioni di dostarlimab a causa della tiroidite.
Si è verificata insufficienza surrenalica in 7 pazienti (1,2%), compresi casi di grado 2 (0,5%) e di grado 3 (0,7%). L'insufficienza surrenalica ha portato all'interruzione di dostarlimab in 1 paziente (0,2%) e si è risolta in 4 pazienti (57,1%).
Nefrite immuno-correlata
Si è verificata nefrite, compresa nefrite tubulo-interstiziale, in 3 pazienti (0,5%), tutti di grado 2. Sono stati necessari corticosteroidi sistemici (prednisone ≥ 40 mg/die o equivalente) in 2 pazienti (66,7%) che avevano manifestato nefrite. La nefrite ha portato all'interruzione di dostarlimab in 1 paziente (0,2%) e si è risolta in tutti e 3 i pazienti.
Eruzione cutanea immuno-correlata
Nei pazienti in trattamento con dostarlimab, si è verificata eruzione cutanea immuno-correlata (eruzione cutanea, eruzione cutanea maculo-papulare, eruzione cutanea maculare, eruzione cutanea pruriginosa, pemfigoide, eruzione da farmaco, tossicità cutanea, eruzione cutanea tossica) in 31 pazienti (5,1%), di cui 9 casi (1,5%) di grado 3. Il tempo mediano all'insorgenza dell'eruzione cutanea è stato di 57 giorni (intervallo (range): da 2 giorni a 1 485 giorni). L'uso di corticosteroidi sistemici (prednisone ≥ 40 mg/die o equivalente) si è reso necessario in 9 pazienti (29,0%) che avevano manifestato eruzione cutanea. L'eruzione cutanea ha portato all'interruzione di dostarlimab in 1 paziente (0,2%) e si è risolta in 24 pazienti (77,4%).
Artralgia immuno-correlata
Si è verificata artralgia immuno-correlata in 34 pazienti (5,6%). Artralgia immuno-correlata di grado 3 è stata segnalata in 5 pazienti (0,8%) in trattamento con dostarlimab. Il tempo mediano all'insorgenza dell'artralgia è stato di 94,5 giorni (intervallo (range): da 1 giorno a 840 giorni). Sono stati necessari corticosteroidi sistemici (prednisone ≥ 40 mg/die o equivalente) in 3 pazienti (8,8%) che avevano manifestato artralgia. L'artralgia ha portato all'interruzione di dostarlimab in 1 paziente (0,2%) e si è risolta in 19 pazienti (55,9%) che avevano manifestato artralgia.
Reazioni da infusione
Si sono verificate reazioni da infusione, compresa ipersensibilità, in 6 pazienti (1,0%), comprese reazioni da infusione di grado 2 (0,3%) e di grado 3 (0,2%). Tutti i pazienti si sono ristabiliti dalle reazioni correlate a infusione.
Immunogenicità
Nello studio GARNET , la presenza di anticorpi anti-farmaco (Anti-Drug Antibodies, ADA) è stata valutata in 315 pazienti in trattamento con dostarlimab e l'incidenza di ADA diretti contro dostarlimab emersi durante il trattamento è risultata pari al 2,5%. Nell'1,3% dei pazienti sono stati rilevati anticorpi neutralizzanti. La co-somministrazione con carboplatino e paclitaxel non ha influenzato l'immunogenicità di dostarlimab. Nello studio RUBY, nelle 225 pazienti trattate con dostarlimab in associazione a carboplatino e paclitaxel e valutabili per presenza di ADA, non vi è stata incidenza di ADA emergenti dal trattamento con dostarlimab o di anticorpi neutralizzanti emergenti dal trattamento.
Nei pazienti che avevano sviluppato ADA non sono emerse evidenze di alterazioni dell'efficacia o della sicurezza di dostarlimab.
Popolazione anziana
Dei 605 pazienti trattati con dostarlimab in monoterapia, il 51,6% aveva un'età inferiore a 65 anni, il 36,9% un'età compresa fra 65 e meno di 75 anni e l'11,5% un'età pari o superiore a 75 anni. Complessivamente non sono state riscontrate differenze nel profilo di sicurezza nei pazienti anziani (≥ 65 anni) rispetto ai soggetti più giovani (< 65 anni).
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l'autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite il sito web dell'Agenzia Italiana del Farmaco, https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Sovradosaggio
Cosa fare se avete preso una dose eccessiva di Jemperli
Se si sospetta un sovradosaggio, la paziente deve essere sottoposta a monitoraggio per individuare segni o sintomi di reazioni o effetti avversi e deve essere istituito un appropriato trattamento sintomatico.
Scadenza
Flaconcino chiuso
3 anni.
Dopo diluizione
Se il medicinale non viene utilizzato immediatamente, la stabilità chimica e fisica in uso è stata dimostrata per 24 ore a una temperatura compresa tra 2°C e 8°C e per 6 ore a temperatura ambiente (fino a 25°C) dal momento della preparazione/diluizione fino al termine della somministrazione.
Conservazione
Conservare in frigorifero (2°C - 8°C).
Non congelare.
Conservare nella confezione originale per proteggere il medicinale dalla luce.
Per le condizioni di conservazione dopo la diluizione vedere paragrafo 6.3.
Foglietto Illustrativo
Fonti Ufficiali
Servizi Avanzati



© 2022 EDRA S.p.A. - P.iva 08056040960
DPO - dpo@lswr.it