
Binocrit
Ultimo aggiornamento: 10/08/2023
Cos'è Binocrit?
Binocrit è un farmaco a base del principio attivo
Epoetina Alfa, appartenente alla categoria degli
Antianemici, eritropoietina e nello specifico
Altri preparati antianemici. E' commercializzato in Italia dall'azienda
Sandoz S.p.A..
Binocrit può essere prescritto con Ricetta RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti.
Binocrit può essere prescritto con Ricetta RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti.
Confezioni
Binocrit 1000 UI/0,5 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,5 ml con disp. ago
Binocrit 10000 UI/1 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 1 ml con disp. ago
Binocrit 2000 UI/1 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 1 ml con disp. ago
Binocrit 20000 UI/0,5 ml sc o iv 1 sir. prer. 0,5 ml c/dispositivo sicurezza per ago
Binocrit 3000 UI/0,3 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,3 ml con disp. ago
Binocrit 30000 UI/0,75 ml sc o ev 1 sir. prer. 0,75 ml c/dispositivo sicurezza per ago
Binocrit 4000 UI/0,4 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,4 ml con disp. ago
Binocrit 40000 UI/1 ml sc o ev 1 siringa prer. 1 ml c/dispositivo sicurezza per ago
Binocrit 5000 UI/0,5 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,5 ml con disp. ago
Binocrit 6000 UI/0,6 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,6 ml con disp. ago
Binocrit 8000 UI/0,8 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,8 ml con disp. ago
Binocrit 10000 UI/1 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 1 ml con disp. ago
Binocrit 2000 UI/1 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 1 ml con disp. ago
Binocrit 20000 UI/0,5 ml sc o iv 1 sir. prer. 0,5 ml c/dispositivo sicurezza per ago
Binocrit 3000 UI/0,3 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,3 ml con disp. ago
Binocrit 30000 UI/0,75 ml sc o ev 1 sir. prer. 0,75 ml c/dispositivo sicurezza per ago
Binocrit 4000 UI/0,4 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,4 ml con disp. ago
Binocrit 40000 UI/1 ml sc o ev 1 siringa prer. 1 ml c/dispositivo sicurezza per ago
Binocrit 5000 UI/0,5 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,5 ml con disp. ago
Binocrit 6000 UI/0,6 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,6 ml con disp. ago
Binocrit 8000 UI/0,8 ml soluz. iniett. in sir. preriemp. uso sc o iv 6 sir. preriemp. 0,8 ml con disp. ago
Informazioni commerciali sulla prescrizione
Titolare: Sandoz GmbH
Concessionario: Sandoz S.p.A.
Ricetta: RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti
Classe: A
Principio attivo: Epoetina Alfa
Gruppo terapeutico: Antianemici, eritropoietina
ATC: B03XA01 - Eritropoietina
Forma farmaceutica: soluzione
Concessionario: Sandoz S.p.A.
Ricetta: RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti
Classe: A
Principio attivo: Epoetina Alfa
Gruppo terapeutico: Antianemici, eritropoietina
ATC: B03XA01 - Eritropoietina
Forma farmaceutica: soluzione
Se sei un professionista, potrai trovare le schede tecniche complete e molto altro nell'area riservata di Codifa.it
Indicazioni
Perché si usa Binocrit? A cosa serve?
Binocrit è indicato per il trattamento dell'anemia sintomatica associata a insufficienza renale cronica (IRC):
- negli adulti e nei bambini di età compresa tra 1 e 18 anni emodializzati e in pazienti adulti sottoposti a dialisi peritoneale (vedere paragrafo 4.4).
- negli adulti con insufficienza renale non ancora dializzati per il trattamento dell'anemia grave, di origine renale, accompagnata da sintomi clinici (vedere paragrafo 4.4).
Binocrit è indicato negli adulti in trattamento chemioterapico per tumori solidi, linfoma maligno o mieloma multiplo e a rischio di trasfusione, come indicato dallo stato generale del paziente (situazione cardiovascolare, anemia preesistente all'inizio della chemioterapia) per il trattamento dell'anemia e la riduzione del fabbisogno trasfusionale.
Binocrit è indicato negli adulti facenti parte di un programma di predonazione autologa per aumentare la produzione di sangue autologo. Il trattamento deve essere effettuato solo in pazienti con anemia moderata (intervallo di concentrazione dell'emoglobina [Hb] compreso tra 10 e 13 g/dL [tra 6,2 e 8,1 mmol/L], senza carenza di ferro), quando le tecniche di risparmio di sangue non siano disponibili o siano insufficienti e l'intervento programmato di chirurgia elettiva maggiore richieda un elevato quantitativo di sangue (4 o più unità di sangue per le donne, 5 o più unità per gli uomini).
Binocrit è indicato negli adulti non sideropenici, ritenuti ad alto rischio di complicanze trasfusionali, prima di un intervento elettivo di chirurgia ortopedica maggiore, per ridurre l'esposizione a trasfusioni di sangue allogenico. Limitare l'uso ai pazienti con anemia moderata (intervallo di concentrazione dell'emoglobina compreso tra 10 e 13 g/dL o tra 6,2 e 8,1 mmol/L) non facenti parte di un programma di predonazione autologa e per i quali si preveda una perdita ematica moderata (900-1.800 mL).
Binocrit è indicato per il trattamento dell'anemia sintomatica (concentrazione emoglobinica ≤ 10 g/dL) negli adulti con sindromi mielodisplastiche (MDS) primarie a rischio basso o intermedio-1, che presentano bassi livelli di eritropoietina sierica (< 200 mU/mL).
Posologia
Come usare Binocrit: Posologia
La terapia con Binocrit deve essere iniziata sotto la supervisione di medici esperti nel trattamento di pazienti con le indicazioni riportate precedentemente.
Posologia
Devono essere valutate tutte le altre cause di anemia (carenza di ferro, folato o vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi e fibrosi del midollo osseo di qualsiasi origine) e devono essere trattate prima di iniziare la terapia con Epoetina Alfa e nel momento in cui si decide di aumentare la dose. Per garantire una risposta ottimale all'epoetina alfa, occorre assicurarsi che esistano depositi di ferro adeguati e, se necessario, somministrare un supplemento di ferro (vedere paragrafo 4.4).
Trattamento dell'anemia sintomatica in pazienti adulti con insufficienza renale cronica
I sintomi e le conseguenze dell'anemia possono variare a seconda di età, sesso e comorbidità mediche; è necessaria una valutazione individuale del decorso clinico e delle condizioni di ogni singolo paziente da parte del medico.
L'intervallo di concentrazione emoglobinica desiderato raccomandato è compreso tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L). Binocrit deve essere somministrato in modo che i valori di emoglobina non aumentino oltre 12 g/dL (7,5 mmol/L). Deve essere evitato un aumento dell'emoglobina superiore a 2 g/dL (1,25 mmol/L) nell'arco di settimane. Se ciò dovesse verificarsi, deve essere effettuato un aggiustamento posologico appropriato.
A causa della variabilità intra-paziente, possono essere occasionalmente osservati valori emoglobinici individuali superiori e inferiori all'intervallo di concentrazione emoglobinica auspicato. La variabilità dell'emoglobina deve essere affrontata tramite la gestione della dose, tenendo conto dell'intervallo di concentrazione emoglobinica compreso tra 10 g/dL (6,2 mmol/L) e 12 g/dL (7,5 mmol/L).
Livelli emoglobinici prolungati superiori a 12 g/dL (7,5 mmol/L) devono essere evitati. Se l'emoglobina aumenta di oltre 2 g/dL (1,25 mmol/L) al mese, o in presenza di livelli emoglobinici prolungati superiori a 12 g/dL (7,5 mmol/L), ridurre la dose di Binocrit del 25%. Se l'emoglobina supera i 13 g/dL (8,1 mmol/L), interrompere la terapia fino a che i valori scendano sotto i 12 g/dL (7,5 mmol/L) e quindi riprendere il trattamento con Binocrit ad una dose inferiore del 25% rispetto alla dose precedente.
I pazienti devono essere attentamente monitorati per assicurarsi che venga usata la dose efficace più bassa approvata di Binocrit per un controllo adeguato dell'anemia e dei sintomi dell'anemia, pur mantenendo una concentrazione emoglobinica inferiore o pari 12 g/dL (7,5 mmol/L).
Usare cautela nell'incremento delle dosi di Binocrit nei pazienti con insufficienza renale cronica. Nei pazienti con scarsa risposta emoglobinica a Binocrit, devono essere prese in considerazione spiegazioni alternative alla base della scarsa risposta (vedere paragrafi 4.4 e 5.1).
Il trattamento con Binocrit consiste di due fasi: la fase di correzione e la fase di mantenimento.
Pazienti adulti emodializzati
Nei pazienti in emodialisi ove sia prontamente disponibile l'accesso endovenoso, è preferibile la somministrazione per via endovenosa.
Fase di correzione
La dose iniziale è di 50 UI/kg, tre volte alla settimana.
Se necessario, aumentare o diminuire la dose di 25 UI/kg (tre volte alla settimana) fino a raggiungere l'intervallo di concentrazione emoglobinica desiderato, compreso tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L) (questo deve avvenire gradualmente ad intervalli di almeno quattro settimane).
Fase di mantenimento
La dose settimanale totale raccomandata è compresa tra 75 UI/kg e 300 UI/kg.
Deve essere effettuato un aggiustamento appropriato della dose per mantenere i valori di emoglobina entro l'intervallo di concentrazione desiderato, compreso tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L).
I pazienti con valori emoglobinici inizialmente molto bassi (< 6 g/dL o < 3,75 mmol/L) possono necessitare di dosi di mantenimento più alte rispetto ai pazienti con anemia iniziale meno grave (> 8 g/dL o > 5 mmol/L).
Pazienti adulti con insufficienza renale non ancora dializzati
Ove non sia prontamente disponibile l'accesso endovenoso, Binocrit può essere somministrato per via sottocutanea.
Fase di correzione
Dose iniziale di 50 UI/kg, 3 volte alla settimana, seguita, se necessario, da incrementi di 25 UI/kg (3 volte alla settimana) fino al raggiungimento del valore desiderato (questo deve avvenire gradualmente ad intervalli di almeno quattro settimane).
Fase di mantenimento
Durante la fase di mantenimento, Binocrit può essere somministrato 3 volte alla settimana oppure, in caso di somministrazione sottocutanea, una volta alla settimana o una volta ogni 2 settimane.
Deve essere effettuato un aggiustamento appropriato della dose e degli intervalli di dosaggio per mantenere i valori di emoglobina al livello desiderato: emoglobina compresa tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L). L'estensione dell'intervallo tra le dosi può richiedere un aumento del dosaggio.
Il dosaggio massimo non deve superare le 150 UI/kg 3 volte alla settimana, 240 UI/kg (fino a un massimo di 20.000 UI) una volta alla settimana, o 480 UI/kg (fino a un massimo di 40.000 UI) una volta ogni 2 settimane.
Pazienti adulti sottoposti a dialisi peritoneale
Ove non sia prontamente disponibile l'accesso endovenoso, Binocrit può essere somministrato per via sottocutanea.
Fase di correzione
La dose iniziale è di 50 UI/kg, due volte alla settimana.
Fase di mantenimento
La dose di mantenimento raccomandata è compresa tra 25 UI/kg e 50 UI/kg, due volte alla settimana, in 2 iniezioni uguali.
Deve essere effettuato un aggiustamento appropriato della dose per mantenere i valori di emoglobina al livello desiderato, compreso tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L).
Trattamento di pazienti adulti con anemia indotta dalla chemioterapia
I sintomi e le conseguenze dell'anemia possono variare a seconda di età, sesso e gravità complessiva della malattia; è necessaria una valutazione individuale del decorso clinico e delle condizioni di ogni singolo paziente da parte del medico.
Binocrit deve essere somministrato ai pazienti anemici (ad es. con concentrazione emoglobinica ≤ 10 g/dL (6,2 mmol/L)).
La dose iniziale è di 150 UI/kg per via sottocutanea, 3 volte alla settimana.
In alternativa, Binocrit può essere somministrato a una dose iniziale di 450 UI/kg per via sottocutanea una volta alla settimana.
Deve essere effettuato un aggiustamento appropriato della dose per mantenere i valori di emoglobina entro l'intervallo di concentrazione desiderato, compreso tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L).
A causa della variabilità intra-paziente, possono essere osservate occasionalmente singole concentrazioni emoglobiniche superiori e inferiori all'intervallo di concentrazione emoglobinica auspicato. Si raccomanda di affrontare la variabilità dell'emoglobina tramite una gestione ottimale della dose, tenendo presente l'intervallo di concentrazione emoglobinica auspicato compreso tra 10 g/dL (6,2 mmol/L) e 12 g/dL (7,5 mmol/L). Concentrazioni emoglobiniche prolungate superiori a 12 g/dL (7,5 mmol/L) devono essere evitate; le linee guida per un adattamento appropriato della dose in caso di concentrazioni emoglobiniche superiori a 12 g/dL (7,5 mmol/L) sono descritte più avanti.
- Se la concentrazione emoglobinica è aumentata di almeno 1 g/dL (0,62 mmol/L) o la conta dei reticolociti è aumentata di ≥ 40.000 cellule/µL sopra il basale dopo quattro settimane di trattamento, si deve mantenere una dose di 150 UI/kg tre volte alla settimana o 450 UI/kg una volta alla settimana.
- Se l'aumento della concentrazione emoglobinica è < 1 g/dL (< 0,62 mmol/L) e la conta dei reticolociti è aumentata di < 40.000 cellule/µL sopra il basale, aumentare la dose a 300 UI/kg tre volte alla settimana. Se, dopo altre quattro settimane di trattamento con 300 UI/kg tre volte alla settimana, l'aumento della concentrazione emoglobinica è ≥ 1 g/dL (≥ 0,62 mmol/L) o la conta dei reticolociti è aumentata di ≥ 40.000 cellule/µL, si deve mantenere la dose di 300 UI/kg tre volte alla settimana.
- Se l'aumento della concentrazione emoglobinica è < 1 g/dL (< 0,62 mmol/L) e la conta dei reticolociti è aumentata di < 40.000 cellule/µL sopra il basale, una risposta è improbabile e il trattamento deve essere interrotto.
Aggiustamento posologico per il mantenimento di una concentrazione emoglobinica compresa tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L)
Se la concentrazione emoglobinica aumenta di oltre 2 g/dL (1,25 mmol/L) al mese, oppure se il livello di concentrazione emoglobinica supera i 12 g/dL (7,5 mmol/L), ridurre la dose di Binocrit di circa il 25-50%.
Se il livello di concentrazione emoglobinica supera i 13 g/dL (8,1 mmol/L), interrompere la terapia fino a che i valori scendano sotto i 12 g/dL (7,5 mmol/L) e quindi riprendere il trattamento con Binocrit ad una dose inferiore del 25% rispetto alla dose precedente.
Il regime posologico raccomandato è riportato nello schema seguente:
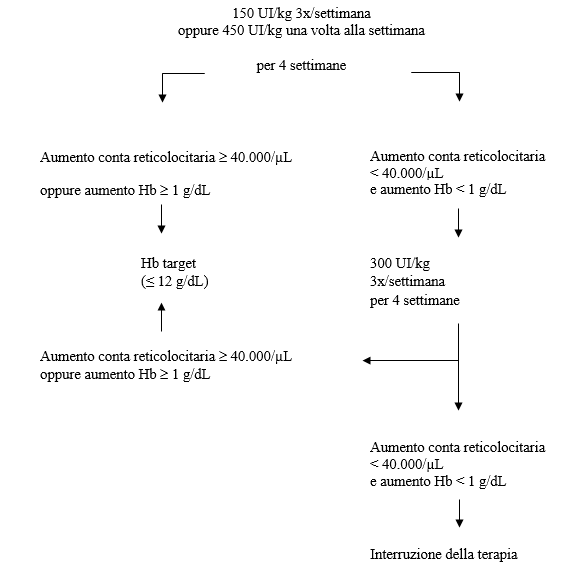
I pazienti devono essere attentamente monitorati per assicurare che venga usata la dose più bassa approvata di agente stimolante l'eritropoiesi (erythropoiesis-stimulating agent, ESA) per un controllo adeguato dei sintomi dell'anemia.
La terapia con epoetina alfa deve proseguire fino a un mese dopo il termine della chemioterapia.
Trattamento di pazienti chirurgici adulti facenti parte di un programma di predonazione autologa
I pazienti con anemia lieve (ematocrito compreso tra 33 e 39%), che necessitino di un predeposito di 4 o più unità di sangue, devono essere trattati con 600 UI/kg di Binocrit per via endovenosa, due volte alla settimana, nelle tre settimane precedenti l'intervento. Binocrit deve essere somministrato dopo che la procedura di donazione sia stata completata.
Trattamento di pazienti adulti in attesa di un intervento elettivo di chirurgia ortopedica maggiore
La dose raccomandata è di 600 UI/kg di Binocrit, somministrata per via sottocutanea una volta alla settimana per tre settimane (giorni -21, -14 e -7) prima dell'intervento e il giorno dell'intervento (giorno 0).
Nei casi in cui, per ragioni mediche, occorra ridurre il tempo fino all'intervento a meno di tre settimane, si devono somministrare giornalmente 300 UI/kg di Binocrit per via sottocutanea per 10 giorni consecutivi prima dell'intervento, il giorno dell'intervento e nei quattro giorni immediatamente successivi.
Se il livello di emoglobina raggiunge o supera i 15 g/dL (9,38 mmol/L) nel periodo preoperatorio, deve essere interrotta la somministrazione di Binocrit e non devono essere somministrati i dosaggi successivi.
Trattamento di pazienti adulti con MDS a rischio basso o intermedio-1
Binocrit deve essere somministrato ai pazienti con anemia sintomatica (ad es. con concentrazione emoglobinica ≤ 10 g/dL (6,2 mmol/L)).
La dose iniziale raccomandata è di 450 UI/kg di Binocrit (la dose totale massima è di 40.000 UI) somministrata per via sottocutanea una volta alla settimana, con almeno 5 giorni di intervallo tra una dose e l'altra.
Devono essere effettuati aggiustamenti appropriati della dose per mantenere le concentrazioni emoglobiniche entro l'intervallo desiderato, compreso tra 10 g/dL e 12 g/dL (tra 6,2 e 7,5 mmol/L). Si raccomanda di valutare la risposta eritroide iniziale da 8 a 12 settimane dopo l'inizio del trattamento. Gli aumenti e le riduzioni della dose devono essere eseguiti a un livello di dose per volta (vedere il diagramma seguente). Deve essere evitata una concentrazione emoglobinica superiore a 12 g/dL (7,5 mmol/L).
Aumento della dose: la dose non deve essere aumentata oltre il massimo di 1.050 UI/kg (dose totale 80.000 UI) alla settimana. Se alla riduzione della dose il paziente smette di rispondere alla terapia o la concentrazione emoglobinica scende di ≥ 1 g/dL, la dose dove essere aumentata di un livello. Devono trascorrere almeno 4 settimane tra un aumento di dose e l'altro.
Mantenimento e riduzione della dose: epoetina alfa deve essere sospesa quando la concentrazione emoglobinica supera il valore di 12 g/dL (7,5 mmol/L). Quando il livello di emoglobina è < 11 g/dL, è possibile riprendere la dose allo stesso livello o a un livello inferiore, in base al giudizio del medico. Prendere in considerazione la riduzione della dose di un livello in caso di un rapido aumento dell'emoglobina (> 2 g/dL nell'arco di 4 settimane).
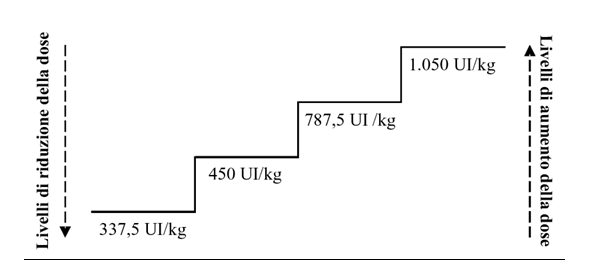
I sintomi e le conseguenze dell'anemia possono variare a seconda di età, sesso e comorbidità mediche; è necessaria una valutazione individuale del decorso clinico e delle condizioni di ogni singolo paziente da parte del medico.
Popolazione pediatrica
Trattamento dell'anemia sintomatica in pazienti con insufficienza renale cronica in emodialisi
I sintomi e le conseguenze dell'anemia possono variare a seconda di età, sesso e comorbidità mediche; è necessaria una valutazione individuale del decorso clinico e delle condizioni di ogni singolo paziente da parte del medico.
Nei pazienti pediatrici, l'intervallo di concentrazione emoglobinica raccomandato è compreso tra 9,5 g/dL e 11 g/dL (tra 5,9 e 6,8 mmol/L). Binocrit deve essere somministrato in modo che i valori di emoglobina non aumentino oltre 11 g/dL (6,8 mmol/L). Deve essere evitato un aumento dell'emoglobina superiore a 2 g/dL (1,25 mmol/L) nell'arco di quattro settimane. Se ciò dovesse verificarsi, deve essere effettuato un aggiustamento posologico appropriato.
I pazienti devono essere attentamente monitorati per assicurarsi che venga usata la dose più bassa approvata di Binocrit per un controllo adeguato dell'anemia e dei sintomi dell'anemia.
Il trattamento con Binocrit consiste di due fasi: la fase di correzione e la fase di mantenimento.
Nei pazienti pediatrici in emodialisi ove sia prontamente disponibile l'accesso endovenoso, è preferibile la somministrazione per via endovenosa.
Fase di correzione
La dose iniziale è di 50 UI/kg per via endovenosa, 3 volte alla settimana.
Se necessario, aumentare o diminuire la dose di 25 UI/kg (tre volte alla settimana) fino a raggiungere l'intervallo di concentrazione emoglobinica desiderato, compreso tra 9,5 g/dL e 11 g/dL (tra 5,9 e 6,8 mmol/L) (questo deve avvenire gradualmente ad intervalli di almeno quattro settimane).
Fase di mantenimento
Deve essere effettuato un aggiustamento appropriato della dose per mantenere i livelli di emoglobina entro l'intervallo di concentrazione desiderato, compreso tra 9,5 g/dL e 11 g/dL (tra 5,9 e 6,8 mmol/L).
In genere, i bambini di peso corporeo inferiore a 30 kg necessitano di dosi di mantenimento più alte rispetto ai bambini di peso corporeo superiore a 30 kg e agli adulti.
I pazienti pediatrici con valori iniziali di emoglobina molto bassi (< 6,8 g/dL o < 4,25 mmol/L) possono aver bisogno di dosi di mantenimento più alte rispetto ai pazienti i cui valori iniziali di emoglobina sono maggiori (> 6,8 g/dL o > 4,25 mmol/L).
Anemia nei pazienti con insufficienza renale cronica prima dell'inizio della dialisi o sottoposti a dialisi peritoneale
La sicurezza e l'efficacia di epoetina alfa nei pazienti con insufficienza renale cronica con anemia prima dell'inizio della dialisi o sottoposti a dialisi peritoneale non sono state stabilite. I dati al momento disponibili per l'uso sottocutaneo di epoetina alfa in queste popolazioni sono riportati nel paragrafo 5.1, ma non può essere fatta alcuna raccomandazione riguardante la posologia.
Trattamento di pazienti pediatrici con anemia indotta dalla chemioterapia
La sicurezza e l'efficacia di epoetina alfa nei pazienti pediatrici sottoposti a chemioterapia non sono state stabilite (vedere paragrafo 5.1).
Trattamento di pazienti chirurigici pediatrici facenti parte di un programma di predonazione autologa
La sicurezza e l'efficacia di epoetina alfa nei soggetti pediatrici non sono state stabilite. Non ci sono dati disponibili.
Trattamento di pazienti pediatrici in attesa di un intervento elettivo di chirurgia ortopedica maggiore
La sicurezza e l'efficacia di epoetina alfa nei soggetti pediatrici non sono state stabilite. Non ci sono dati disponibili.
Modo di somministrazione
Precauzioni che devono essere prese prima della manipolazione o della somministrazione del medicinale.
Prima dell'uso, lasciare riposare la siringa di Binocrit finché non raggiunge la temperatura ambiente. Solitamente sono necessari 15-30 minuti.
Come per tutti gli altri prodotti iniettabili, verificare che la soluzione non contenga particelle e non presenti alterazioni del colore. Binocrit è un prodotto sterile, privo di conservanti, monouso. Somministrare la quantità richiesta.
Trattamento dell'anemia sintomatica in pazienti adulti con insufficienza renale cronica
Nei pazienti con insufficienza renale cronica ove sia regolarmente disponibile l'accesso endovenoso (pazienti emodializzati) è preferibile la somministrazione di Binocrit per via endovenosa.
Ove non sia prontamente disponibile l'accesso endovenoso, (pazienti non ancora dializzati e pazienti sottoposti a dialisi peritoneale) Binocrit può essere somministrato tramite iniezione sottocutanea.
Trattamento di pazienti adulti con anemia indotta dalla chemioterapia
Binocrit deve essere somministrato tramite iniezione sottocutanea.
Trattamento di pazienti chirurgici adulti facenti parte di un programma di predonazione autologa
Binocrit deve essere somministrato per via endovenosa.
Trattamento di pazienti adulti in attesa di un intervento elettivo di chirurgia ortopedica maggiore
Binocrit deve essere somministrato tramite iniezione sottocutanea.
Trattamento di pazienti adulti con MDS a rischio basso o intermedio-1
Binocrit deve essere somministrato tramite iniezione sottocutanea.
Trattamento dell'anemia sintomatica in pazienti pediatrici con insufficienza renale cronica in emodialisi
Nei pazienti pediatrici con insufficienza renale cronica ove sia regolarmente disponibile l'accesso endovenoso (pazienti emodializzati) è preferibile la somministrazione di Binocrit per via endovenosa.
Somministrazione endovenosa
Somministrare per almeno uno - cinque minuti, a seconda della dose totale. Nei pazienti emodializzati può essere somministrato un bolo durante la seduta dialitica attraverso un accesso venoso adatto nella linea di dialisi. Alternativamente, l'iniezione può essere somministrata al termine della seduta dialitica attraverso il tubo dell'ago fistola, seguita da 10 mL di soluzione salina isotonica per sciacquare il tubo e garantire l'iniezione del prodotto in circolo (vedere Posologia, “Pazienti adulti emodializzati“).
Nei pazienti che reagiscono al trattamento con sintomi simil-influenzali è preferibile una somministrazione più lenta (vedere paragrafo 4.8).
Non somministrare Binocrit tramite infusione endovenosa o in associazione con altri medicinali in soluzione (fare riferimento al paragrafo 6.6 per ulteriori informazioni).
Somministrazione sottocutanea
Non superare il volume massimo di 1 mL in ogni sede d'iniezione. Per iniettare volumi maggiori, servirsi di più sedi d'iniezione.
Somministrare l'iniezione negli arti o nella parete addominale anteriore.
Nei casi in cui il medico stabilisca che il paziente o un suo assistente possa somministrare con sicurezza ed efficacia Binocrit per via sottocutanea in modo autonomo, è necessario fornire indicazioni sul dosaggio e sul metodo di somministrazione corretti.
Graduazioni in rilievo
La siringa contiene graduazioni in rilievo per provvedere alla somministrazione di una parte della dose (vedere paragrafo 6.6). Tuttavia il prodotto è esclusivamente monouso. Da ogni siringa si deve prelevare una sola dose di Binocrit.
Le “Istruzioni per l'autoiniezione di Binocrit“ si trovano in calce al foglio illustrativo.
Controindicazioni
Quando non dev'essere usato Binocrit
- Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
- Non somministrare Binocrit o un'altra eritropoietina ai pazienti che sviluppano un'aplasia specifica della serie rossa (PRCA) in seguito al trattamento con una qualsiasi eritropoietina (vedere paragrafo 4.4).
- Ipertensione non controllata.
- Tutte le controindicazioni associate ai programmi di predonazione di sangue autologo devono essere rispettate nei pazienti a cui è fornita un'integrazione di Binocrit.
L'uso di Binocrit nei pazienti in attesa di un intervento elettivo di chirurgia ortopedica maggiore, che non partecipano ad un programma di predonazione autologa, è controindicato nei pazienti con gravi patologie vascolari coronariche, arteriose periferiche, carotidee o cerebrali, compresi i pazienti con infarto miocardico recente o accidente cerebrovascolare.
- Pazienti chirurgici che, per qualsiasi ragione, non possano ricevere una profilassi antitrombotica adeguata.
Avvertenze speciali e precauzioni di impiego
Cosa serve sapere prima di prendere Binocrit
Considerazioni generali
Nei pazienti trattati con Epoetina Alfa, la pressione arteriosa deve essere attentamente monitorata e controllata, secondo necessità. L'epoetina alfa deve essere usata con cautela in presenza di ipertensione non trattata, trattata in misura inadeguata o scarsamente controllabile. Può essere necessario aggiungere medicinali o aumentare la dose della terapia antipertensiva. Interrompere il trattamento con epoetina alfa se la pressione arteriosa non può essere controllata.
Anche durante il trattamento con epoetina alfa di pazienti con pressione arteriosa precedentemente normale o bassa, si sono manifestate crisi ipertensive con encefalopatia e convulsioni richiedenti l'intervento immediato di un medico e la terapia intensiva. Deve essere prestata particolare attenzione a cefalee trafittive improvvise di tipo simil-emicranico, che possono essere un segno premonitore (vedere paragrafo 4.8).
L'epoetina alfa deve essere usata con cautela in pazienti con epilessia, convulsioni all'anamnesi o condizioni mediche associate a una predisposizione all'attività convulsiva, come infezioni del SNC e metastasi cerebrali.
L'epoetina alfa deve essere usata con cautela in pazienti con insufficienza epatica cronica. La sicurezza dell'epoetina alfa non è stabilita in pazienti con disfunzione epatica.
È stata osservata un'aumentata incidenza di eventi trombotici vascolari (TVE) nei pazienti trattati con ESA (vedere paragrafo 4.8). Questi comprendono trombosi ed embolie venose e arteriose (tra cui alcuni casi con esito fatale), come trombosi venosa profonda, emboli polmonari, trombosi retinica e infarto miocardico. Inoltre, sono stati riportati accidenti cerebrovascolari (comprendenti infarto cerebrale, emorragia cerebrale e attacchi ischemici transitori).
Il rischio segnalato di questi TVE deve essere valutato con attenzione in rapporto ai benefici attesi dal trattamento con epoetina alfa, in particolare nei pazienti con fattori di rischio preesistenti per i TVE, comprendenti obesità e TVE all'anamnesi (per es. trombosi venosa profonda, embolia polmonare e accidente cerebrovascolare).
In tutti i pazienti, i livelli di emoglobina devono essere sottoposti a stretto monitoraggio, a causa del rischio potenzialmente più elevato di eventi tromboembolici ed esito fatale nel caso i pazienti vengano trattati a livelli di emoglobina superiori all'intervallo di concentrazione dell'indicazione.
Durante il trattamento con epoetina alfa può verificarsi un moderato aumento dose-dipendente della conta piastrinica nell'ambito del range normale. Tale aumento regredisce durante il proseguimento della terapia. Inoltre, sono stati riportati casi di trombocitemia al di sopra del range normale. Si raccomanda di sottoporre la conta piastrinica a regolare monitoraggio durante le prime otto settimane di terapia.
Devono essere valutate tutte le altre cause di anemia (carenza di ferro, folato o vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi e fibrosi del midollo osseo di qualsiasi origine) e devono essere trattate prima di iniziare la terapia con epoetina alfa e nel momento in cui si decide di aumentare la dose. Nella maggior parte dei casi, i valori di ferritina nel siero si riducono contemporaneamente all'aumento dell'ematocrito. Per garantire una risposta ottimale all'epoetina alfa, occorre assicurarsi che esistano depositi di ferro adeguati e, se necessario, somministrare un supplemento di ferro (vedere paragrafo 4.2):
- Nei pazienti con insufficienza renale cronica, si raccomanda la somministrazione di ferro (ferro elementare, da 200 a 300 mg/die per via orale negli adulti e da 100 a 200 mg/die per via orale nei soggetti pediatrici) se i livelli sierici di ferritina sono inferiori a 100 ng/mL.
- Nei pazienti oncologici, si raccomanda la somministrazione di ferro (ferro elementare, da 200 a 300 mg/die per via orale) se la saturazione della transferrina è inferiore al 20%.
- Nei pazienti facenti parte di un programma di predonazione autologa, la somministrazione di ferro (ferro elementare, 200 mg/die per via orale) deve avvenire diverse settimane prima di iniziare il predeposito autologo, in modo da formare abbondanti depositi di ferro prima dell'inizio della terapia con epoetina alfa, e per l'intera durata della terapia con epoetina alfa.
- Nei pazienti in attesa di un intervento elettivo di chirurgia ortopedica maggiore, la somministrazione di ferro (ferro elementare, 200 mg/die per via orale) deve avvenire per l'intera durata della terapia con epoetina alfa. Se possibile, la somministrazione di ferro deve iniziare prima dell'inizio della terapia con epoetina alfa, in modo da formare depositi di ferro adeguati.
Molto raramente, è stata osservata la comparsa o l'esacerbazione della porfiria in pazienti trattati con epoetina alfa. L'epoetina alfa deve essere usata con cautela nei pazienti con porfiria.
In associazione al trattamento con epoetina, sono state segnalate reazioni avverse cutanee severe (SCAR), incluse la sindrome di Stevens-Johnson (SJS) e la necrolisi tossica epidermica (TEN), che possono essere fatali o rappresentare un rischio per la vita. Sono stati osservati casi più severi con epoetine a lunga durata d'azione.
Al momento della prescrizione, i pazienti devono essere informati in merito ai segni e ai sintomi e si deve attuare un attento monitoraggio al fine di verificare il manifestarsi di potenziali reazioni cutanee. Se si manifestano segni e sintomi riconducibili a queste reazioni, la somministrazione di Binocrit deve essere immediatamente interrotta e si deve prendere in considerazione un trattamento alternativo.
Se il paziente ha sviluppato una reazione cutanea severa come SJS o TEN a causa dell'uso di Binocrit, il trattamento con Binocrit non dovrà mai essere ripreso per quel paziente.
Per migliorare la tracciabilità degli agenti stimolanti l'eritropoiesi (ESA), il nome e il numero di lotto dell'agente somministrato devono essere registrati (oppure indicati) in modo inequivocabile nella documentazione sanitaria del paziente.
I pazienti devono passare da un ESA a un altro esclusivamente sotto supervisione adeguata.
Aplasia specifica della serie rossa (PRCA)
È stata riportata PRCA mediata da anticorpi dopo mesi o anni di trattamento con epoetina alfa. Sono stati riportati anche casi in pazienti con epatite C trattati con interferone e ribavirina, in presenza di terapia concomitante con ESA. L'epoetina alfa non è approvata per il trattamento dell'anemia associata ad epatite C.
Nei pazienti nei quali si osserva improvvisamente una mancata efficacia della terapia, definita da un calo dell'emoglobina (1-2 g/dL o 0,62-1,25 mmol/L al mese) con aumento del fabbisogno trasfusionale, deve essere determinata la conta reticolocitaria e devono essere analizzate le tipiche cause di una mancata risposta (carenza di ferro, folato o vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi e fibrosi del midollo osseo di qualsiasi origine).
In caso di riduzione paradossa dell'emoglobina e insorgenza di anemia grave associata a basse conte reticolocitarie, il trattamento con epoetina alfa deve essere interrotto e devono essere determinati gli anticorpi anti-eritropoietina. Deve essere preso in considerazione anche un esame del midollo osseo per un'eventuale diagnosi di PRCA.
Non devono essere avviate altre terapie con ESA a causa del rischio di reazione crociata.
Trattamento dell'anemia sintomatica in pazienti adulti e pediatrici con insufficienza renale cronica
Nei pazienti con insufficienza renale cronica trattati con epoetina alfa, i livelli di emoglobina devono essere misurati regolarmente fino al raggiungimento di un livello stabile e, successivamente, a intervalli periodici.
Nei pazienti con insufficienza renale cronica, l'aumento dell'emoglobina deve corrispondere approssimativamente a 1 g/dL (0,62 mmol/L) al mese e non deve superare i 2 g/dL (1,25 mmol/L) al mese, per ridurre al minimo il rischio di un peggioramento dell'ipertensione.
Nei pazienti con insufficienza renale cronica, la concentrazione emoglobinica di mantenimento non deve superare il limite superiore dell'intervallo di concentrazione emoglobinica, come raccomandato nel paragrafo 4.2. Negli studi clinici è stato osservato un aumento del rischio di decesso e di eventi cardiovascolari gravi in caso di somministrazione di ESA per ottenere un livello di concentrazione emoglobinica superiore a 12 g/dL (7,5 mmol/L).
Studi clinici controllati non hanno mostrato benefici significativi attribuibili alla somministrazione di epoetine una volta che la concentrazione emoglobinica abbia superato i livelli necessari per controllare i sintomi dell'anemia ed evitare le trasfusioni di sangue.
Deve essere prestata cautela nell'incremento delle dosi di Binocrit nei pazienti con insufficienza renale cronica, perché dosi cumulative elevate di epoetina possono essere associate a un aumento del rischio di mortalità e di gravi eventi cardiovascolari e cerebrovascolari. Nei pazienti con scarsa risposta emoglobinica alle epoetine devono essere prese in considerazione spiegazioni alternative alla base della scarsa risposta (vedere paragrafi 4.2 e 5.1).
I pazienti con insufficienza renale cronica trattati con epoetina alfa per via sottocutanea devono essere sottoposti a un monitoraggio periodico per verificare la perdita di efficacia, definita come assenza o riduzione della risposta al trattamento con epoetina alfa in pazienti che in precedenza rispondevano a tale terapia. Questa condizione è caratterizzata da un calo prolungato dell'emoglobina nonostante un incremento del dosaggio di epoetina alfa (vedere paragrafo 4.8).
Alcuni pazienti che utilizzano intervalli tra le dosi di epoetina alfa più estesi (superiori a una volta alla settimana) potrebbero non riuscire a mantenere livelli emoglobinici adeguati (vedere paragrafo 5.1) e potrebbero necessitare di un aumento della dose di epoetina alfa. I livelli di emoglobina devono essere monitorati con regolarità.
Nei pazienti in emodialisi si sono verificate trombosi dello shunt, particolarmente nei pazienti con tendenza all'ipotensione o con complicazioni a livello delle fistole arterovenose (ad es. stenosi, aneurismi ecc.). In questi pazienti si raccomandano la revisione precoce dello shunt e una profilassi antitrombotica, ad esempio con acido acetilsalicilico.
In casi isolati è stata osservata iperpotassiemia, sebbene non sia stato stabilito un legame causale. Nei pazienti con insufficienza renale cronica devono essere monitorati gli elettroliti del siero. In presenza di un livello di potassio nel siero elevato o crescente, oltre a un trattamento appropriato dell'iperpotassiemia, deve essere valutata l'eventualità di interrompere la somministrazione di epoetina alfa fino alla correzione del livello di potassio nel siero.
Per via dell'aumento dell'ematocrito, durante la terapia con epoetina alfa è spesso necessario un aumento della dose di eparina in corso di emodialisi. Se l'eparinizzazione non è ottimale è possibile che si verifichi un'occlusione del sistema dialitico.
In base alle informazioni attualmente disponibili, la correzione dell'anemia con epoetina alfa in pazienti adulti con insufficienza renale non ancora dializzati non accelera la progressione dell'insufficienza renale.
Trattamento dei pazienti con anemia indotta da chemioterapia
Nei pazienti oncologici trattati con epoetina alfa, i livelli di emoglobina devono essere misurati regolarmente fino al raggiungimento di un livello stabile e, successivamente, a intervalli periodici.
Le epoetine sono fattori di crescita che stimolano soprattutto la produzione di eritrociti. I recettori per l'eritropoietina possono essere espressi sulla superficie di una varietà di cellule tumorali. Come per tutti i fattori di crescita, esiste la preoccupazione che le epoetine possano stimolare la crescita di tumori. Non può escludersi il ruolo degli ESA sulla progressione tumorale o sulla riduzione della sopravvivenza senza progressione. In studi clinici controllati, l'uso di epoetina alfa e altri ESA è stato associato a una riduzione del controllo locoregionale del tumore o a una riduzione della sopravvivenza generale:
- un ridotto controllo locoregionale in pazienti con cancro avanzato del distretto testa-collo trattati con radioterapia, se somministrati per ottenere un livello di concentrazione emoglobinica superiore a 14 g/dL (8,7 mmol/L),
- una riduzione della sopravvivenza generale e un aumento dei decessi attribuiti alla progressione del tumore a 4 mesi in pazienti con carcinoma mammario metastatico trattate con chemioterapia, se somministrati per ottenere un intervallo di concentrazione emoglobinica di 12-14 g/dL (7,5-8,7 mmol/L),
- un aumento del rischio di decesso se somministrati per ottenere un livello di concentrazione emoglobinica di 12 g/dL (7,5 mmol/L) in pazienti con neoplasie maligne attive, non trattati né con chemioterapia né con radioterapia. L'uso di ESA non è indicato in questa popolazione di pazienti.
- è stato osservato tramite un'analisi primaria un aumento del 9% del rischio di progressione della malattia (PD) o decesso nel gruppo trattato con epoetina alfa e terapia standard (SOC) e un aumento del rischio del 15%, che non può essere escluso dal punto di vista statistico, nelle pazienti con carcinoma mammario metastatico trattate con chemioterapia, quando somministrati per ottenere un intervallo di concentrazione emoglobinica di 10-12 g/dL (6,2-7,5 mmol/L).
Sulla base di quanto riportato sopra, in alcune condizioni cliniche la trasfusione di sangue deve essere il trattamento preferito per la gestione dell'anemia nei pazienti affetti da neoplasia. La decisione di effettuare un trattamento con eritropoietina ricombinante deve essere basata sulla valutazione del rapporto beneficio-rischio con il coinvolgimento del singolo paziente e deve prendere in considerazione lo specifico contesto clinico. I fattori che devono essere considerati in questa valutazione devono includere il tipo di tumore e il relativo stadio, il grado di anemia, l'aspettativa di vita, l'ambiente nel quale il paziente è trattato e le preferenze del paziente stesso (vedere paragrafo 5.1).
Nei pazienti oncologici in chemioterapia, si deve tenere presente l'intervallo di 2-3 settimane tra la somministrazione di ESA e la comparsa degli eritrociti indotti dall'eritropoietina nella valutazione della appropriatezza della terapia con epoetina alfa (pazienti a rischio di trasfusione).
Pazienti chirurgici facenti parte di programmi di predonazione autologa
Devono essere rispettate tutte le avvertenze e precauzioni speciali relative ai programmi di predonazione autologa; in particolare, la sostituzione di volume deve essere eseguita di routine.
Pazienti in attesa di un intervento elettivo di chirurgia ortopedica maggiore
Seguire sempre le pratiche di buona gestione del sangue nel perioperatorio.
I pazienti in attesa di un intervento elettivo di chirurgia ortopedica maggiore devono ricevere un'adeguata profilassi antitrombotica, in quanto, nei pazienti chirurgici, possono verificarsi eventi trombotici e vascolari, specialmente nei pazienti con patologia cardiovascolare di base. Si deve prestare, inoltre, particolare cautela nei pazienti predisposti a sviluppare DVT (trombosi venosa profonda). Inoltre, nei pazienti con emoglobina al basale > 13 g/dL (> 8,1 mmol/L), non può essere esclusa la possibilità che il trattamento con epoetina alfa possa essere associato ad un rischio aumentato di eventi trombotici/vascolari post-operatori. Pertanto, l'epoetina alfa non deve essere impiegata nei pazienti con emoglobina basale > 13 g/dL (> 8,1 mmol/L).
Eccipienti
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per siringa preriempita, cioè è praticamente “senza sodio“.
Interazioni con altri medicinali e altre forme di interazione
Quali farmaci o alimenti possono modificare l'effetto di Binocrit
Non esiste alcuna evidenza che indichi che il trattamento con epoetina alfa alteri il metabolismo di altri medicinali.
I medicinali che riducono l'eritropoiesi possono ridurre la risposta all'epoetina alfa.
Dal momento che la ciclosporina viene legata dagli eritrociti, esiste la possibilità di interazione tra medicinali. Se l'epoetina alfa viene somministrata contemporaneamente alla ciclosporina, devono essere sottoposti a monitoraggio i livelli ematici di ciclosporina e deve essere modificata la dose di ciclosporina con l'aumento dell'ematocrito.
Non esiste alcuna evidenza che indichi un'interazione tra epoetina alfa e il fattore stimolante le colonie di granulociti (G-CSF) o il fattore stimolante le colonie di granulociti e macrofagi (GM-CSF) relativamente alla differenziazione o proliferazione ematologica in campioni bioptici tumorali in vitro.
In pazienti adulte con carcinoma mammario metastatico, la co-somministrazione sottocutanea di 40.000 UI/mL di epoetina alfa con 6 mg/kg di trastuzumab non ha avuto alcun effetto sulla farmacocinetica di trastuzumab.
Fertilità, gravidanza e allattamento
Gravidanza
I dati relativi all'uso di Epoetina Alfa in donne in gravidanza non esistono o sono in numero limitato. Gli studi sugli animali hanno mostrato una tossicità riproduttiva (vedere paragrafo 5.3).
Pertanto, l'epoetina alfa deve essere usata in gravidanza solo se il potenziale beneficio supera il potenziale rischio per il feto. L'uso di epoetina alfa non è raccomandato nelle pazienti chirurgiche in gravidanza facenti parte di un programma di predonazione autologa.
Allattamento
Non è noto se l'epoetina alfa esogena sia escreta nel latte materno. L'epoetina alfa deve essere usata con cautela nelle donne che allattano. Si deve decidere se interrompere l'allattamento o interrompere la terapia/astenersi dalla terapia con epoetina alfa tenendo in considerazione il beneficio dell'allattamento per il bambino e il beneficio della terapia per la donna.
L'uso di epoetina alfa non è raccomandato nelle pazienti chirurgiche in allattamento facenti parte di un programma di predonazione autologa.
Fertilità
Non vi sono studi volti a determinare l'effetto potenziale dell'epoetina alfa sulla fertilità maschile o femminile.
Effetti sulla capacità di guidare veicoli e sull'uso di macchinari
Non sono stati effettuati studi sulla capacità di guidare veicoli e sull'uso di macchinari. Binocrit non altera o altera in modo trascurabile la capacità di guidare veicoli o di usare macchinari.
Effetti indesiderati
Quali sono gli effetti collaterali di Binocrit
Riassunto del profilo di sicurezza
La reazione avversa al farmaco più frequente durante il trattamento con Epoetina Alfa è l'aumento dose-dipendente della pressione arteriosa o il peggioramento di un'ipertensione preesistente. La pressione arteriosa deve essere monitorata, in particolare all'inizio della terapia (vedere paragrafo 4.4).
Le reazioni avverse al farmaco più frequenti osservate negli studi clinici con epoetina alfa sono le seguenti: diarrea, nausea, vomito, piressia e cefalea. La malattia simil-influenzale può manifestarsi in particolare all'inizio del trattamento.
Una congestione del tratto respiratorio, comprendente eventi di congestione del tratto respiratorio superiore, congestione nasale e nasofaringite, è stata segnalata in studi con intervalli estesi tra le dosi in pazienti adulti affetti da insufficienza renale e non ancora sottoposti a dialisi.
È stata osservata un'aumentata incidenza di eventi trombotici vascolari (TVE) nei pazienti trattati con ESA (vedere paragrafo 4.4).
Tabella delle reazioni avverse
Su un totale di 3.417 soggetti inclusi in 25 studi randomizzati, in doppio cieco, controllati verso placebo o terapia standard, il profilo di sicurezza complessivo dell'epoetina alfa è stato valutato in 2.094 soggetti anemici. Sono stati inclusi 228 soggetti con insufficienza renale cronica trattati con epoetina alfa in 4 studi sull'insufficienza renale cronica (2 studi in pre-dialisi [N = 131 soggetti esposti con insufficienza renale cronica] e 2 in dialisi [N = 97 soggetti esposti con insufficienza renale cronica]; 1.404 soggetti oncologici esposti in 16 studi sull'anemia dovuta a chemioterapia; 147 soggetti esposti in 2 studi sulla donazione di sangue autologo; 213 soggetti esposti in 1 studio nel periodo perioperatorio e 102 soggetti esposti in 2 studi sulle MDS. Le reazioni avverse al farmaco riferite da ≥ 1% dei soggetti trattati con epoetina alfa in questi studi sono riportate nella tabella in basso.
Stima della frequenza: molto comune (≥ 1/10); comune (≥ 1/100, < 1/10); non comune (≥ 1/1.000, < 1/100); raro (≥1/10.000, < 1/1.000); molto raro (< 1/10.000), non nota (la frequenza non può essere definita sulla base dei dati disponibili).
|
Classificazione per sistemi e organi (SOC) secondo MedDRA
|
Reazione avversa (livello di termine preferito)
|
Frequenza
|
|
Patologie del sistema emolinfopoietico
|
Aplasia specifica della serie rossa3, trombocitemia
|
Raro
|
|
Disturbi del metabolismo e della nutrizione
|
Iperkaliemia1
|
Non comune
|
|
Disturbi del sistema immunitario
|
Ipersensibilità3
|
Non comune
|
|
Reazione anafilattica3
|
Raro
|
|
|
Patologie del sistema nervoso
|
Cefalea
|
Comune
|
|
Convulsioni
|
Non comune
|
|
|
Patologie vascolari
|
Ipertensione, trombosi venosa e arteriosa2
|
Comune
|
|
Crisi ipertensiva3
|
Non nota
|
|
|
Patologie respiratorie, toraciche e mediastiniche
|
Tosse
|
Comune
|
|
Congestione delle vie respiratorie
|
Non comune
|
|
|
Patologie gastrointestinali
|
Diarrea, nausea, vomito
|
Molto comune
|
|
Patologie della cute e del tessuto sottocutaneo
|
Eruzione cutanea
|
Comune
|
|
Orticaria3
|
Non comune
|
|
|
Edema angioneurotico3
|
Non nota
|
|
|
Patologie del sistema muscoloscheletrico e del tessuto connettivo
|
Artralgia, dolore osseo, mialgia, dolore a un arto
|
Comune
|
|
Patologie congenite, familiari e genetiche
|
Porfiria acuta3
|
Raro
|
|
Patologie sistemiche e condizioni relative alla sede di somministrazione
|
Piressia
|
Molto comune
|
|
Brividi, malattia simil-influenzale, reazioni in sede di iniezione, edema periferico
|
Comune
|
|
|
Medicinale inefficace3
|
Non nota
|
|
|
Esami diagnostici
|
Anticorpo anti-eritropoietina positivo
|
Raro
|
|
1 Comune nella dialisi
2 Include eventi arteriosi e venosi, fatali e non fatali, come trombosi venosa profonda, emboli polmonari, trombosi retinica, trombosi arteriosa (infarto miocardico incluso), accidenti cerebrovascolari (infarto cerebrale ed emorragia cerebrale inclusi), attacchi ischemici transitori, trombosi dello shunt (apparecchiature per dialisi incluse) e trombosi in aneurismi di shunt arterovenosi
3 Discusso nel sottoparagrafo in basso e/o nel paragrafo 4.4.
|
||
Descrizione di reazioni avverse selezionate
Sono state riportate reazioni di ipersensibilità comprendenti casi di eruzione cutanea (orticaria inclusa), reazioni anafilattiche ed edema angioneurotico (vedere paragrafo 4.4).
Anche durante il trattamento con epoetina alfa di pazienti con pressione arteriosa precedentemente normale o bassa, si sono manifestate crisi ipertensive con encefalopatia e convulsioni richiedenti l'intervento immediato di un medico e la terapia intensiva. Deve essere prestata particolare attenzione a cefalee trafittive improvvise di tipo simil-emicranico, che possono essere un segno premonitore (vedere paragrafo 4.4).
In associazione al trattamento con epoetina, sono state segnalate reazioni avverse cutanee severe (SCAR), incluse la sindrome di Stevens-Johnson (SJS) e la necrolisi tossica epidermica (TEN), che possono essere fatali o rappresentare un rischio per la vita (vedere paragrafo 4.4).
Molto raramente è stata riportata aplasia specifica della serie rossa mediata da anticorpi (in < 1/10.000 casi per anno paziente) dopo mesi o anni di trattamento con epoetina alfa (vedere paragrafo 4.4). Sono stati riportati più casi con la via di somministrazione sottocutanea (s.c.) rispetto alla via e.v.
Pazienti adulti con MDS a rischio basso o intermedio-1
Nello studio randomizzato, in doppio cieco, controllato verso placebo, multicentrico, 4 soggetti (4,7%) hanno manifestato TVE (morte improvvisa, ictus ischemico, embolia e flebite). Tutti i TVE si sono verificati nel gruppo trattato con epoetina alfa e nelle prime 24 settimane dello studio. Tre sono stati TVE confermati, mentre nel caso rimanente (morte improvvisa), l'evento tromboembolico non è stato confermato. Due soggetti presentavano fattori di rischio significativi (fibrillazione atriale, insufficienza cardiaca e tromboflebite).
Popolazione pediatrica con insufficienza renale cronica in emodialisi
L'esposizione di pazienti pediatrici con insufficienza renale cronica in emodialisi in studi clinici e nell'esperienza post-marketing è limitata. In questa popolazione non è stata segnalata alcuna reazione avversa specifica per i soggetti pediatrici che non sia citata nella tabella precedente o che non sia coerente con la patologia di base.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l'autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite l'Agenzia Italiana del Farmaco, sito web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Sovradosaggio
Cosa fare se avete preso una dose eccessiva di Binocrit
Il margine terapeutico dell'Epoetina Alfa è molto ampio. Un sovradosaggio di epoetina alfa può causare effetti che rappresentano un'estensione degli effetti farmacologici dell'ormone. In presenza di livelli di emoglobina eccessivamente elevati è possibile ricorrere alla flebotomia. In caso di necessità si deve ricorrere a trattamenti di supporto addizionali.
Scadenza
2 anni
Conservazione
Conservare e trasportare in frigorifero (2°C - 8°C). Questo intervallo di temperatura deve essere osservato strettamente fino alla somministrazione al paziente.
Per l'uso ambulatoriale, il medicinale può essere prelevato dal frigorifero, senza riporvelo nuovamente, per un periodo massimo di 3 giorni a temperatura non superiore a 25 °C. Se il medicinale non è stato utilizzato al termine di tale periodo, deve essere eliminato.
Non congelare o agitare.
Conservare nella confezione originale per proteggere il medicinale dalla luce.
Farmaci Equivalenti
I farmaci equivalenti di
Binocrit a base di
Epoetina Alfa sono:
Eprex
Foglietto Illustrativo
Fonti Ufficiali
Servizi Avanzati



© 2022 EDRA S.p.A. - P.iva 08056040960
DPO - dpo@lswr.it