

Cos'è Neorecormon?
Neorecormon è un farmaco a base del principio attivo Epoetina Beta , appartenente alla categoria degli Antianemici, eritropoietina e nello specifico Altri preparati antianemici. E' commercializzato in Italia dall'azienda Roche S.p.A. .
Neorecormon può essere prescritto con RicettaRNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti .
Neorecormon può essere prescritto con Ricetta
Confezioni
Neorecormon 2000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 3000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 4000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 5000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 6000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon sottoc. ev 1 siringa 10.000 UI/0,6 ml
Neorecormon sottoc. ev 1 siringa 30000 UI/0,6 ml
Neorecormon 3000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 4000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 5000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon 6000 UI/0,3 ml soluzione iniettabile 6 siringhe preriempite e 6 aghi
Neorecormon sottoc. ev 1 siringa 10.000 UI/0,6 ml
Neorecormon sottoc. ev 1 siringa 30000 UI/0,6 ml
Informazioni commerciali sulla prescrizione
Titolare: Roche Registration GmbH
Concessionario:Roche S.p.A.
Ricetta:RNRL - medicinali soggetti a prescrizione medica limitativa, da rinnovare volta per volta, vendibili al pubblico su prescrizione di centri ospedalieri o di specialisti
Classe:A
Principio attivo:Epoetina Beta
Gruppo terapeutico:Antianemici, eritropoietina
ATC:B03XA01 - Eritropoietina
Forma farmaceutica: soluzione
Concessionario:
Ricetta:
Classe:
Principio attivo:
Gruppo terapeutico:
ATC:
Forma farmaceutica: soluzione
Se sei un professionista, potrai trovare le schede tecniche complete e molto altro nell'area riservata di Codifa.it
Indicazioni
Perché si usa Neorecormon? A cosa serve?
NeoRecormon è indicato per:
- trattamento dell'anemia sintomatica associata ad insufficienza renale cronica in pazienti adulti e pediatrici.
- prevenzione dell'anemia dei neonati prematuri con un peso alla nascita compreso tra 750 e 1500 g e con un periodo di gestazione inferiore a 34 settimane.
- trattamento dell'anemia sintomatica in pazienti adulti con tumore non mieloide sottoposti a chemioterapia.
- incrementare la quantità di sangue autologo in pazienti facenti parte di un programma di predonazione. Il suo uso in questa indicazione deve essere valutato in rapporto all'aumentato rischio di eventi tromboembolici. Il trattamento deve essere riservato solo a pazienti con anemia di grado moderato (emoglobina 10 - 13 g/dl [6,21 - 8,07 mmol/l], in assenza di carenza di ferro) se le procedure di conservazione non sono disponibili o sono insufficienti quando l'intervento elettivo di chirurgia maggiore richiede un notevole volume di sangue (4 o più unità di sangue per le donne o 5 o più unità per gli uomini). Vedere paragrafo 5.1.
Posologia
Come usare Neorecormon: Posologia
La terapia con NeoRecormon deve essere iniziata da medici esperti nelle sopracitate indicazioni. Essendo state segnalate in casi isolati reazioni anafilattoidi, si raccomanda di somministrare la prima dose sotto il controllo del medico.
Posologia
Trattamento dell'anemia sintomatica in pazienti adulti e pediatrici con insufficienza renale cronica
Sintomi e sequele dell'anemia possono variare in funzione dell'età, del sesso e del carico complessivo della malattia; è necessario che il decorso clinico e le condizioni del singolo paziente siano valutati dal medico. NeoRecormon deve essere somministrato per via sottocutanea o endovenosa per aumentare l'emoglobina fino a un livello non superiore a 12 g/dl (7,45 mmol/l). La via sottocutanea è da preferirsi in pazienti non sottoposti a emodialisi per evitare punture alle vene periferiche. In caso di somministrazione endovenosa, la soluzione deve essere iniettata in circa 2 minuti, nei pazienti emodializzati attraverso la fistola arterovenosa alla fine della dialisi.
In considerazione della variabilità intrapaziente, possono essere occasionalmente rilevati, in un paziente, singoli valori di emoglobina superiori e inferiori al livello di emoglobina desiderato. La variabilità dell'emoglobina deve essere gestita attraverso l'aggiustamento della dose, in riferimento ad un intervallo target di emoglobina tra 10 g/dl (6,21 mmol/l) e 12 g/dl (7,45 mmol/l). Si deve evitare un livello prolungato di emoglobina superiore a 12 g/dl (7,45 mmol/l); le indicazioni per una appropriata correzione del dosaggio per quando vengono osservati valori di emoglobina superiori a 12 g/dl (7,45 mmol/l) sono riportate di seguito.
Si deve evitare un incremento dell'emoglobina maggiore di 2 g/dl (1,25 mmol/l) nell'arco di quattro settimane. Se ciò si verifica, si deve procedere ad una appropriata correzione del dosaggio, come indicato. Se l'entità dell'aumento dell'emoglobina è superiore a 2 g/dl (1,25 mmol/l) in un mese o se il livello di emoglobina aumenta e si avvicina a 12 g/dl (7,45 mmol/l), la dose deve essere ridotta del 25% circa. Se il livello di emoglobina continua a crescere, si deve interrompere la terapia fino a quando il livello di emoglobina comincia a diminuire, punto al quale la terapia deve essere ricominciata a una dose inferiore del 25% circa rispetto a quella somministrata in precedenza.
I pazienti devono essere monitorati attentamente per garantire che venga utilizzata la più bassa dose efficace autorizzata di NeoRecormon per controllare adeguatamente i sintomi dell'anemia mantenendo una concentrazione di emoglobina inferiore o uguale a 12g / dl (7.45 mmol/l).
Si deve usare cautela nell'incremento delle dosi di NeoRecormon in pazienti con insufficienza renale cronica. Nei pazienti con una scarsa risposta emoglobinica a NeoRecormon devono essere prese in considerazione spiegazioni alternative per tale scarsa risposta (vedere paragrafi 4.4 e 5.1).
In presenza di ipertensione o di patologie cardiovascolari, cerebrovascolari, o vascolari periferiche, l'incremento settimanale di Hb e il valore massimo di Hb da raggiungere devono essere determinati su base individuale, considerando il quadro clinico.
Il trattamento con NeoRecormon è diviso in due fasi:
- Fase di correzione
- Somministrazione sottocutanea:
Il dosaggio iniziale è di 3 x 20 UI/kg di peso corporeo alla settimana. Se l'aumento di Hb non fosse adeguato (< 0,25 g/dl/settimana), il dosaggio può essere aumentato ogni 4 settimane di 3 x 20 UI/kg alla settimana.
Il dosaggio settimanale può essere ripartito in somministrazioni giornaliere. - Somministrazione endovenosa:
Il dosaggio iniziale è di 3 x 40 UI/kg alla settimana. Il dosaggio può essere aumentato, dopo 4 settimane, a 80 UI/kg - tre volte alla settimana - e con ulteriori incrementi di 20 UI/kg, se necessario, tre volte alla settimana, ad intervalli mensili.
Per entrambe le vie di somministrazione, non si deve superare la dose massima di 720 UI/kg alla settimana.
- Fase di mantenimento
Per mantenere il livello dell'Hb entro un range compreso tra 10 e 12 g/dl, la dose è inizialmente ridotta alla metà di quella precedentemente somministrata. Successivamente, la dose viene adattata su base individuale per paziente (dose di mantenimento) ad intervalli di una o due settimane.
In caso di somministrazione sottocutanea, la dose totale settimanale può essere somministrata con un'unica iniezione settimanale o può essere divisa in tre o sette dosi settimanali. Pazienti stabili con un regime di singola somministrazione settimanale possono passare ad una somministrazione ogni due settimane. In tal caso potrebbe essere necessario incrementare la dose.
I risultati emersi dagli studi clinici in bambini hanno dimostrato che, mediamente, più giovani sono i pazienti, più elevate sono le dosi di NeoRecormon necessarie. Tuttavia ci si deve attenere allo schema posologico raccomandato, in quanto la risposta individuale non può essere prevista.
Il trattamento con NeoRecormon è di norma a lungo termine. Tuttavia, qualora fosse necessario, può essere interrotto in ogni momento. I dati relativi allo schema posologico una volta alla settimana si basano su studi clinici della durata di 24 settimane di terapia.
Prevenzione dell'anemia del prematuro
La soluzione viene somministrata per via sottocutanea ad una dose di 3 x 250 UI/kg di peso corporeo alla settimana. I bambini prematuri che, all'inizio del trattamento con NeoRecormon, abbiano già ricevuto trasfusioni, rispondono meno alla terapia rispetto ai bambini non trasfusi. La durata del trattamento raccomandata è di 6 settimane.
Trattamento dell'anemia sintomatica indotta da chemioterapia in pazienti con tumori
NeoRecormon deve essere somministrato per via sottocutanea in pazienti anemici (ad es. con concentrazione di emoglobina ≤ 10 g/dl (6,21 mmol/l)). Sintomi e sequele dell'anemia possono variare in funzione dell'età, del sesso e del carico complessivo della malattia; è necessario che il decorso clinico e le condizioni del singolo paziente siano valutati dal medico.
La dose settimanale può essere fornita attraverso una unica somministrazione settimanale oppure attraverso 3-7 iniezioni settimanali.
La dose iniziale raccomandata è di 30.000 UI alla settimana (pari a circa 450 UI/kg di peso corporeo alla settimana, in base al peso corporeo del paziente).
In considerazione della variabilità intrapaziente, possono essere occasionalmente rilevati, in un paziente, singoli valori di emoglobina superiori e inferiori al livello di emoglobina desiderato. La variabilità dell'emoglobina deve essere gestita attraverso l'aggiustamento della dose, in riferimento ad un intervallo target di emoglobina tra 10 g/dl (6,21 mmol/l) e 12 g/dl (7,45 mmol/l). Si deve evitare un livello prolungato di emoglobina superiore a 12 g/dl (7,45 mmol/l); le indicazioni per una appropriata correzione del dosaggio per quando vengono osservati valori di emoglobina superiori a 12 g/dl (7,45 mmol/l) sono riportate di seguito.
Se, dopo 4 settimane di terapia, il valore emoglobinico è aumentato di almeno 1 g/dl (0,62 mmol/l), deve essere mantenuta la dose corrente. Se il valore emoglobinico non è aumentato di almeno 1 g/dl (0,62 mmol/l), si può considerare il raddoppio della dose settimanale. Se, dopo 8 settimane di terapia, il valore emoglobinico non è aumentato di almeno 1 g/dl (0,62 mmol/l), è improbabile che si verifichi una risposta, e il trattamento deve essere interrotto.
La terapia deve essere continuata per 4 settimane dopo la fine della chemioterapia.
La dose massima non deve superare 60.000 UI a settimana.
Una volta raggiunto l'obiettivo terapeutico per un paziente, la dose deve essere ridotta dal 25 al 50% al fine di mantenere l'emoglobina a quel livello.
Occorre considerare di effettuare una appropriata titolazione della dose.
Se l'emoglobina supera 12 g/dl (7,45 mmol/l), la dose deve essere ridotta del 25 - 50% circa. Il trattamento con NeoRecormon deve essere interrotto temporaneamente se i livelli di emoglobina superano 13 g/dl (8,1 mmol/l). Quando il livello dell'emoglobina scende a un valore uguale o inferiore a 12 g/dl (7,45 mmol/l), la terapia deve essere ripresa a una dose inferiore di circa il 25% rispetto alla precedente.
Se l'aumento dell'emoglobina è maggiore di 2 g/dl (1,3 mmol/l) in 4 settimane, la dose deve essere ridotta dal 25 al 50%.
I pazienti devono essere monitorati attentamente per garantire che venga utilizzata la più bassa dose autorizzata di NeoRecormon per controllare adeguatamente i sintomi dell'anemia.
Trattamento per incrementare la quantità di sangue autologo
La soluzione viene somministrata per via endovenosa in circa 2 minuti o per via sottocutanea.
NeoRecormon viene somministrato due volte alla settimana per 4 settimane. Nei casi in cui il valore di ematocrito sia tale da rendere possibile la donazione di sangue (Ht ≥ 33%), NeoRecormon viene somministrato al termine della donazione di sangue.
Durante l'intero ciclo di trattamento, l'ematocrito non deve superare il valore di 48%.
Il dosaggio deve essere determinato dall'équipe chirurgica su base individuale per ogni paziente in funzione della quantità di sangue predonato richiesta e della riserva endogena eritrocitaria:
- La quantità richiesta di sangue predonato dipende dalla perdita di sangue prevista, dall'uso delle procedure impiegate per la conservazione del sangue e dalle condizioni fisiche del paziente.
Tale quantità di sangue dovrebbe essere quella prevista essere sufficiente per evitare trasfusioni omologhe di sangue.
La quantità di sangue predonato richiesta è espressa in unità, dove una unità nel nomogramma corrisponde a 180 ml di eritrociti. - La capacità di donare il sangue dipende principalmente dal volume ematico del paziente e dal valore basale di ematocrito. Entrambe le variabili determinano la riserva endogena eritrocitaria che può essere calcolata in accordo alla seguente formula:
Riserva eritrocitaria endogena = volume ematico [ml] x (Ht - 33): 100
donne: volume ematico [ml] = 41 [ml/kg] x peso corporeo [kg] + 1200 [ml]
uomini: volume ematico [ml] = 44 [ml/kg] x peso corporeo [kg] + 1600 [ml] (peso corporeo ≥ 45 kg)
L'indicazione per intraprendere il trattamento con NeoRecormon e per determinare la singola dose, deve essere ricavata in base alla quantità di sangue predonato richiesta e alla riserva eritrocitaria endogena in accordo ai grafici seguenti.
|
Donne Quantità di sangue predonato richiesta [unità]
|
Uomini Quantità di sangue predonato richiesta [unità] |
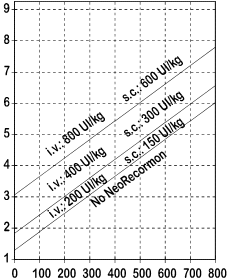 |
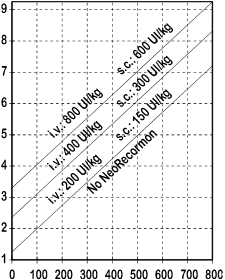 |
| Riserva endogena eritrocitaria [ml] |
Riserva endogena eritrocitaria [ml]
|
La singola dose, così determinata, viene somministrata 2 volte alla settimana per 4 settimane. La dose massima non deve superare 1600 UI/kg di peso corporeo alla settimana per la somministrazione endovenosa o 1200 UI/kg di peso corporeo alla settimana per la somministrazione sottocutanea.
Modo di somministrazione
La siringa pre-riempita di NeoRecormon è pronta per l'uso. Possono essere iniettate solo soluzioni che siano chiare o leggermente opalescenti, incolori e praticamente prive di particelle visibili. NeoRecormon in siringa pre-riempita è un medicinale sterile ma senza conservanti. Per nessun motivo si deve somministrare più di una dose per siringa; il medicinale è esclusivamente per dose singola.
Controindicazioni
Quando non dev'essere usato Neorecormon
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
Ipertensione scarsamente controllata.
Nell'indicazione “incremento della quantità di sangue autologo“: infarto miocardico o ictus nel mese precedente il trattamento, angina pectoris instabile, aumentato rischio di trombosi venose profonde come anamnesi di malattia venosa tromboembolica.
Avvertenze speciali e precauzioni di impiego
Cosa serve sapere prima di prendere Neorecormon
Tracciabilità
Al fine di migliorare la tracciabilità dei medicinali biologici, il nome e il numero di lotto del medicinale somministrato devono essere chiaramente registrati.
NeoRecormon deve essere usato con cautela in presenza di anemia refrattaria con eccesso di blasti in trasformazione, epilessia, trombocitosi, insufficienza epatica cronica. Deficit di acido folico e di vitamina B12 devono essere compensati poiché riducono l'efficacia di NeoRecormon.
Si deve usare cautela nell'incremento delle dosi di NeoRecormon in pazienti con insufficienza renale cronica poiché dosi cumulative elevate di epoetina possono essere associate ad un aumentato rischio di mortalità e di gravi eventi cardiovascolari e cerebrovascolari. Nei pazienti con una scarsa risposta emoglobinica alle epoetine, devono essere prese in considerazione spiegazioni alternative per tale scarsa risposta (vedere paragrafi 4.2 e 5.1).
Per assicurare un'efficace eritropoiesi, lo stato marziale deve essere valutato in tutti i pazienti prima e durante il trattamento, e può essere necessaria una terapia integrativa con ferro, condotta in accordo alle linee guida relative alla terapia.
Un grave sovraccarico di alluminio conseguente il trattamento dell'insufficienza renale, può compromettere l'efficacia di NeoRecormon.
L'indicazione per il trattamento con NeoRecormon di pazienti con nefrosclerosi, non ancora sottoposti a dialisi, deve essere stabilita individualmente, dal momento che non può essere esclusa un'eventuale accelerazione nella progressione della insufficienza renale.
Aplasia pura della serie rossa (PRCA)
È stata segnalata aplasia pura della serie rossa (PRCA) causata da anticorpi neutralizzanti anti-eritropoietina in associazione alla terapia con eritropoietina, incluso NeoRecormon. È stato dimostrato che questi anticorpi reagiscono in modo crociato con tutte le proteine eritropoietiche e i pazienti con sospetta o confermata presenza di anticorpi neutralizzanti anti-eritropoietina non devono passare al trattamento con NeoRecormon (vedere paragrafo 4.8).
PRCA in pazienti affetti da epatite C
Una diminuzione paradossa dell'emoglobina e lo sviluppo di anemia severa associata a una bassa conta reticolocitaria deve indurre a interrompere il trattamento con epoetina e ad eseguire test anticorpali anti-eritropoietina. Casi sono stati riportati in pazienti con epatite C trattati con interferone e ribavirina, contemporaneamente all'utilizzo di epoetine. Le epoetine non sono approvate per il trattamento dell'anemia associata a epatite C.
Monitoraggio della pressione arteriosa
Può verificarsi un aumento della pressione arteriosa o un aggravamento di uno stato ipertensivo esistente soprattutto in caso di aumento rapido dell'ematocrito. Questi aumenti della pressione arteriosa possono essere trattati con farmaci. Se gli aumenti della pressione arteriosa non possono essere controllati con i farmaci si consigli un'interruzione temporanea della terapia con NeoRecormon. In particolare, all'inizio della terapia è raccomandato il regolare monitoraggio della pressione arteriosa, inclusi gli intervalli tra le dialisi. Possono verificarsi crisi ipertensive con sintomi simil-encefalopatia che richiedono l'immediato intervento di un medico e cure mediche intensive. Come possibile segnale d'allarme, particolare attenzione dovrebbe essere prestata a improvvise emicranie lancinanti.
In associazione al trattamento con epoetina, sono state segnalate reazioni avverse cutanee severe (SCAR), incluse la sindrome di Stevens-Johnson (SJS) e la necrolisi tossica epidermica (TEN), che possono essere fatali o rappresentare un rischio per la vita (vedere paragrafo 4.8). Sono stati osservati casi più severi con epoetine a lunga durata d'azione. Al momento della prescrizione, i pazienti devono essere informati in merito ai segni e ai sintomi e si deve attuare un attento monitoraggio al fine di verificare potenziali reazioni cutanee. Se si manifestano segni e sintomi riconducibili a queste reazioni, la somministrazione di deve essere immediatamente sospesa e si dovrà prendere in considerazione un trattamento alternativo. Se il paziente ha sviluppato una reazione cutanea severa come SJS o TEN a causa dell'uso di NeoRecormon, il trattamento con agenti stimolanti l'eritropoiesi (ESA) non dovrà mai essere ripreso per quel paziente.
Insufficienza Renale Cronica
In pazienti con insufficienza renale cronica, si può osservare un moderato aumento dose-dipendente nella conta piastrinica all'interno dell'intervallo di normalità durante il trattamento con NeoRecormon, soprattutto in seguito a somministrazione endovenosa. Questo fenomeno regredisce con il proseguimento della terapia. Si raccomanda di controllare regolarmente la conta piastrinica durante le prime 8 settimane di terapia.
Concentrazione dell'emoglobina
In pazienti con insufficienza renale cronica, la concentrazione di mantenimento dell'emoglobina non deve superare il limite superiore del livello target di emoglobina, raccomandato nel paragrafo 4.2. Negli studi clinici è stato osservato un rischio aumentato di morte e di gravi eventi cardiovascolari o eventi cerebrovascolari che includono l'ictus quando sono stati somministrati ESA, per raggiungere un livello target di emoglobina superiore a 12 g/dl (7,45 mmol/l).
Studi clinici controllati non hanno dimostrato benefici significativi attribuibili alla somministrazione di epoetine, quando la concentrazione di emoglobina viene aumentata oltre il livello necessario a controllare i sintomi dell'anemia e ad evitare trasfusioni ematiche.
Nei neonati prematuri si può manifestare un lieve aumento nella conta piastrinica, in particolar modo fino al 12°-14° giorno di vita; deve pertanto essere controllata regolarmente la conta piastrinica.
Effetto sulla crescita tumorale
Le eritropoietine sono fattori di crescita che stimolano principalmente la produzione di globuli rossi. I recettori dell'eritropoietina possono essere espressi sulla superficie di diverse cellule tumorali. Come avviene con tutti i fattori di crescita, c'è la possibilità che le eritropoietine possano stimolare la crescita di tumori. In diversi studi controllati le epoetine non hanno dimostrato di migliorare la sopravvivenza globale o di determinare una riduzione del rischio di progressione tumorale in pazienti con anemia associata a cancro.
In studi clinici controllati, l'impiego di NeoRecormon e di altri ESA ha dimostrato:
- una riduzione del tempo alla progressione tumorale in pazienti con tumore della testa e del collo allo stadio avanzato sottoposti a radioterapia, se trattati per raggiungere un livello target di emoglobina superiore a 14 g/dl (8,69 mmol/l);
- una ridotta sopravvivenza globale e un aumento delle morti attribuiti a progressione della malattia a 4 mesi, in pazienti con tumore mammario metastatico in chemioterapia quando trattati per raggiungere un livello target di emoglobina tra 12 e 14 g/dl (7,45-8,69 mmol/l);
- un aumentato rischio di morte in pazienti con tumore maligno attivo non sottoposti a chemioterapia né radioterapia, quando trattati per raggiungere un livello target di emoglobina di 12 g/dl (7,45 mmol/l). L'impiego degli ESA non è indicato in questa popolazione di pazienti.
Sulla base di quanto riportato sopra, in alcune condizioni cliniche la trasfusione di sangue deve essere il trattamento preferito per la gestione dell'anemia nei pazienti affetti da neoplasia. La decisione di somministrare eritropoietine ricombinanti deve essere basata sulla valutazione del rapporto beneficio-rischio con il coinvolgimento del singolo paziente e deve prendere in considerazione lo specifico contesto clinico. I fattori che devono essere considerati in questa valutazione devono includere il tipo di tumore e il relativo stadio, il grado di anemia, l'aspettativa di vita, l'ambiente nel quale il paziente è trattato e le preferenze del paziente stesso (vedere paragrafo 5.1).
Può verificarsi un aumento della pressione arteriosa che può essere trattato con farmaci. Si raccomanda quindi di monitorare la pressione arteriosa, in particolare nella fase iniziale del trattamento nei pazienti con tumore.
Anche la conta piastrinica e il valore di emoglobina devono essere controllati ad intervalli regolari nei pazienti con tumore.
In pazienti coinvolti in un programma di predonazione di sangue autologo si può manifestare un aumento della conta piastrinica, prevalentemente all'interno dell'intervallo di normalità. Perciò si raccomanda di misurare la conta piastrinica almeno una volta alla settimana in questi pazienti. Se il numero delle piastrine è superiore a 150 x 109/l o supera i valori normali, il trattamento con NeoRecormon deve essere interrotto.
Nei neonati prematuri, non si può escludere un rischio potenziale di retinopatia causata da eritropoietina, pertanto, si deve prestare particolare attenzione, e la decisione di trattare un neonato prematuro deve essere bilanciata considerando il beneficio potenziale ed il rischio di tale trattamento, nonché le opzioni alternative disponibili.
In pazienti con insufficienza renale cronica, un aumento della dose di eparina durante l'emodialisi è spesso richiesto nel corso del trattamento con NeoRecormon a causa di un incremento del valore di ematocrito. È possibile che si verifichi un'occlusione del sistema dialitico se l'eparinizzazione non è ottimale.
In pazienti con insufficienza renale cronica a rischio di trombosi dello shunt artero-venoso devono essere prese in considerazione una revisione precoce dello shunt ed una profilassi antitrombotica attraverso, ad esempio, la somministrazione di acido acetilsalicilico.
Durante la terapia con NeoRecormon i livelli sierici di potassio e di fosfato devono essere regolarmente controllati. In un numero limitato di pazienti uremici in trattamento con NeoRecormon è stato riportato un incremento del potassio, sebbene il rapporto di causalità non sia stato stabilito. Nel caso in cui si osservi un valore elevato, o in aumento, di potassio si deve prendere in considerazione l'interruzione del trattamento con NeoRecormon fino al ripristino dei valori normali.
Per l'impiego di NeoRecormon in un programma di predonazione autologa, si devono seguire le linee guida ufficiali sulla donazione di sangue, in particolare:
- possono sottoporsi a donazioni soltanto pazienti con un valore di Ht ≥ 33% (emoglobina ≥ 11 g/dl [6.83 mmol/l]);
- speciale cautela deve essere osservata in pazienti di peso inferiore a 50 kg;
- il volume di un singolo prelievo non deve superare approssimativamente il 12% del volume totale stimato di sangue del paziente.
Il trattamento deve essere riservato a pazienti per i quali sia considerato di particolare importanza evitare trasfusioni di sangue omologo e sia stato valutato il rapporto rischio/beneficio derivante da trasfusioni omologhe.
Uso improprio
L'uso improprio da parte di soggetti sani può indurre un eccessivo aumento dell'ematocrito. Ciò può essere associato a complicazioni a carico del sistema cardiovascolare con rischio per la vita.
Eccipienti
NeoRecormon in siringhe preriempite contiene fino a 0,3 mg di fenilalanina/siringa come eccipiente. Pertanto, si deve tenere conto di ciò nei pazienti affetti da gravi forme di fenilchetonuria.
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per siringa, cioè essenzialmente “senza sodio“.
Interazioni con altri medicinali e altre forme di interazione
Quali farmaci o alimenti possono modificare l'effetto di Neorecormon
I risultati clinici ottenuti fino ad ora non hanno evidenziato alcuna interazione di NeoRecormon con altri medicinali.
Esperimenti effettuati su animali hanno dimostrato che epoetina beta non potenzia l'effetto mielotossico di medicinali citostatici quali etoposide, cisplatino, ciclofosfamide e fluorouracile.
Fertilità, gravidanza e allattamento
Gravidanza
Non esistono dati relativi all'uso di NeoRecormon nelle donne in gravidanza. È necessario prestare particolare attenzione nel prescrivere il medicinale a donne in stato di gravidanza.
Allattamento
Non è noto se la beta epoetina sia escreta nel latte materno. La decisione se continuare/interrompere l'allattamento al seno o continuare/interrompere la terapia con beta epoetina deve essere presa tenendo in considerazione il beneficio dell'allattamento al seno per il bambino e il beneficio della terapia con beta epoetina per la donna.
Fertilità
Gli studi su animali non indicano effetti dannosi diretti o indiretti su gravidanza, sviluppo embrionale/fetale, parto o sviluppo post-natale (vedere paragrafo 5.3).
Effetti sulla capacità di guidare veicoli e sull'uso di macchinari
NeoRecormon non altera la capacità di guidare veicoli o di usare macchinari.
Effetti indesiderati
Quali sono gli effetti collaterali di Neorecormon
Sintesi del profilo di sicurezza
Sulla base dei risultati degli studi clinici che hanno coinvolto 1725 pazienti, si prevede che circa l'8% dei pazienti trattati con NeoRecormon sperimenti reazioni avverse.
Pazienti anemici con insufficienza renale cronica
La più frequente reazione avversa durante il trattamento con NeoRecormon è rappresentata da un aumento della pressione arteriosa o dall'aggravamento di uno stato ipertensivo preesistente, specialmente nei casi di aumento rapido del valore di ematocrito (vedere paragrafo 4.4). Crisi ipertensive accompagnate da sintomi analoghi a quelli dell'encefalopatia (ad es. mal di testa e stato confusionale, disturbi sensorio-motori - come disturbi della parola e della deambulazione - fino a convulsioni tonico-cloniche) possono anche insorgere in pazienti normotesi o ipotesi (vedere paragrafo 4.4).
Possono verificarsi trombosi dello shunt in pazienti che hanno la tendenza all'ipotensione o le cui fistole arterovenose presentano complicanze (ad es. stenosi, aneurismi), vedere paragrafo 4.4. In molti casi si osserva una riduzione del valore di ferritina sierica associata ad un incremento dell'ematocrito (vedere paragrafo 4.4). Inoltre, in casi isolati, sono stati osservati aumenti transitori dei livelli sierici di potassio e fosfato (vedere paragrafo 4.4).
In casi isolati è stata riportata aplasia della serie rossa causata da anticorpi neutralizzanti anti- eritropoietina, associata a terapia con NeoRecormon. Nel caso sia diagnosticata aplasia della serie rossa causata da anticorpi anti-eritropoietina, la terapia con NeoRecormon deve essere interrotta e i pazienti non devono essere trattati con un'altra proteina eritropoietica (vedere paragrafo 4.4).
Le reazioni avverse sono elencate nella Tabella 1 sottostante.
Pazienti con tumori
L'ipertensione e la cefalea correlati al trattamento con Epoetina Beta, che possono essere trattati con farmaci, sono comuni (vedere paragrafo 4.4).
In alcuni pazienti si è osservata una riduzione dei parametri sierici del ferro (vedere paragrafo 4.4).
Gli studi clinici hanno mostrato una frequenza più elevata di eventi tromboembolici nei pazienti con tumore trattati con NeoRecormon rispetto ai pazienti dei gruppi di controllo trattati con placebo o non trattati. Nei pazienti trattati con NeoRecormon questa incidenza è del 7% rispetto al 4% nel gruppo di controllo; questo non è associato con nessun aumento della mortalità per eventi tromboembolici rispetto al gruppo di controllo.
Le reazioni avverse sono elencate nella Tabella 2 sottostante.
Pazienti in un programma di predonazione autologa di sangue
Nei pazienti sottoposti ad un programma di predonazione autologa di sangue è stata riportata una incidenza di eventi tromboembolici lievemente superiore. Tuttavia non poteva essere stabilito un rapporto causale con la terapia con NeoRecormon.
In studi controllati verso placebo, il deficit temporaneo di ferro è più pronunciato nel gruppo trattato con NeoRecormon rispetto al controllo (vedere paragrafo 4.4).
Le reazioni avverse sono elencate nella Tabella 3 sottostante.
Sono state segnalate reazioni avverse cutanee severe (SCAR), incluse la sindrome di Stevens-Johnson (SJS) e la necrolisi tossica epidermica (TEN), che possono essere fatali o rappresentare un rischio per la vita (vedere paragrafo 4.4).
Classificazione degli eventi avversi
Gli eventi avversi sono elencati secondo la classificazione MeDRA per sistemi e organi e per categoria di frequenza.
Le categorie di frequenza sono definite mediante la seguente convenzione:
molto comune (>1/10); comune (>1%, <10%); non comune (>0,1%, <1%); raro (>0,01%, <0,1%); molto raro (<1/10.000); non nota (la frequenza non può essere definita sulla base dei dati disponibili).
Tabella 1: Reazioni avverse attribuite al trattamento con NeoRecormon in studi clinici controllati condotti su pazienti affetti da MRC
|
Classificazione sistemica organica
|
Reazione avversa
|
Frequenza
|
|
Patologie vascolari
|
Ipertensione
|
Comune
|
|
Crisi ipertensive
|
Non comune
|
|
|
Patologie del sistema nervoso
|
Cefalea
|
Comune
|
|
Patologie del sistema emolinfopoietico
|
Trombosi dell'accesso vascolare
|
Raro
|
|
Trombocitosi
|
Molto raro
|
Tabella 2: Reazioni avverse attribuite al trattamento con NeoRecormon in studi clinici controllati condotti su pazienti oncologici
|
Classificazione sistemica organica
|
Reazione avversa
|
Frequenza
|
|
Patologie vascolari
|
Ipertensione
|
Comune
|
|
Patologie del sistema emolinfopoietico
|
Eventi tromboembolici
|
Comune
|
|
Patologie del sistema nervoso
|
Cefalea
|
Comune
|
Tabella 3: Reazioni avverse attribuite al trattamento con NeoRecormon in studi clinici controllati condotti su pazienti in un programma di predonazione autologa di sangue
|
Classificazione sistemica organica
|
Reazione avversa
|
Frequenza
|
|
Patologie del sistema nervoso
|
Cefalea
|
Comune
|
Bambini prematuri
Una riduzione dei valori di ferritina sierica è molto comune (vedere paragrafo 4.4).
Descrizione delle reazioni avverse selezionate
Sono state osservate raramente reazioni cutanee correlate al trattamento con epoetina beta, quali eruzione cutanea, prurito, orticaria o reazioni nella sede di iniezione. In casi molto rari, sono state osservate reazioni anafilattoidi correlate al trattamento con epoetina beta. Tuttavia, in studi clinici controllati non si è osservata una più elevata incidenza delle reazioni di ipersensibilità.
In casi molti rari, in particolare all'inizio del trattamento, sono stati riportati sintomi di tipo influenzale correlati al trattamento con epoetina beta, quali febbre, brividi, mal di testa, dolore agli arti, malessere e/o dolore alle ossa. Queste reazioni erano di grado lieve o moderato e scomparivano nell'arco di un paio di ore o giorni.
Dati provenienti da uno studio controllato con epoetina alfa o darbepoetina alfa, hanno riportato un'incidenza di ictus come comune.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette che si verificano dopo l'autorizzazione del medicinale è importante, in quanto permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. Agli operatori sanitari è richiesto di segnalare qualsiasi reazione avversa sospetta tramite il sistema nazionale di segnalazione riportato all'indirizzo https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse.
Sovradosaggio
Cosa fare se avete preso una dose eccessiva di Neorecormon
La finestra terapeutica di NeoRecormon è molto ampia. Non sono stati osservati sintomi di sovradosaggio anche a concentrazioni sieriche molto elevate.
Scadenza
2 anni.
Conservazione
Conservare in frigorifero (2°C − 8°C).
Tenere la siringa preriempita nell'imballaggio esterno per tenerla al riparo dalla luce.
Per l'uso ambulatoriale, il paziente può togliere il medicinale dal frigorifero e conservarlo a temperatura ambiente (non superiore ai 25°C) per un singolo periodo di 3 giorni al massimo.
Elenco degli eccipienti
Urea,
Sodio cloruro,
Polisorbato 20,
Sodio fosfato monobasico diidrato,
Sodio fosfato dibasico dodecaidrato,
Calcio cloruro diidrato,
Glicina,
L-Leucina,
L-Isoleucina,
L-Treonina,
L-Acido glutammico,
L-Fenilalanina,
Acqua per preparazioni iniettabili.
